The Research
Pathophysiology of Dyt1-Tor1a dystonia in mice is mediated by spinal neural circuit dysfunction
Dystonia, a neurological disorder defined by abnormal postures and disorganized movements, is considered to be a neural circuit disorder with dysfunction arising within and between multiple brain regions. Given that spinal neural circuits constitute the final pathway for motor control, we sought to determine their contribution to this movement disorder. Focusing on the most common inherited form of dystonia in humans, DYT1-TOR1A, we generated a conditional knockout of the torsin family 1 member A (Tor1a) gene in the mouse spinal cord and dorsal root ganglia (DRG). We found that these mice recapitulated the phenotype of the human condition, developing early-onset generalized torsional dystonia. Motor signs emerged early in the mouse hindlimbs before spreading caudo-rostrally to affect the pelvis, trunk, and forelimbs throughout postnatal maturation. Physiologically, these mice bore the hallmark features of dystonia, including spontaneous contractions at rest and excessive and disorganized contractions, including cocontractions of antagonist muscle groups, during voluntary movements. Spontaneous activity, disorganized motor output, and impaired monosynaptic reflexes, all signs of human dystonia, were recorded from isolated mouse spinal cords from these conditional knockout mice. All components of the monosynaptic reflex arc were affected, including motor neurons. Given that confining the Tor1a conditional knockout to DRG did not lead to early-onset dystonia, we conclude that the pathophysiological substrate of this mouse model of dystonia lies in spinal neural circuits. Together, these data provide new insights into our current understanding of dystonia pathophysiology.
Blockade of M4 muscarinic receptors on striatal cholinergic interneurons normalizes striatal dopamine release in a mouse model of TOR1A dystonia
Anthony M.Downs, Yuping Donsante, H.A.Jinnah, Ellen J.Hess
Abstract
Trihexyphenidyl (THP), a non-selective muscarinic receptor (mAChR) antagonist, is commonly used for the treatment of dystonia associated with TOR1A, otherwise known as DYT1 dystonia. A better understanding of the mechanism of action of THP is a critical step in the development of better therapeutics with fewer side effects. We previously found that THP normalizes the deficit in striatal dopamine (DA) release in a mouse model of TOR1A dystonia (Tor1a+/ΔE knockin (KI) mice), revealing a plausible mechanism of action for this compound, considering that abnormal DA neurotransmission is consistently associated with many forms of dystonia. However, the mAChR subtype(s) that mediate the rescue of striatal dopamine release remain unclear. In this study we used a combination of pharmacological challenges and cell-type specific mAChR conditional knockout mice of either sex to determine which mAChR subtypes mediate the DA release-enhancing effects of THP. We determined that THP acts in part at M4 mAChR on striatal cholinergic interneurons to enhance DA release in both Tor1a+/+ and Tor1a+/ΔE KI mice. Further, we found that the subtype selective M4 antagonist VU6021625 recapitulates the effects of THP. These data implicate a principal role for M4 mAChR located on striatal cholinergic interneurons in the mechanism of action of THP and suggest that subtype selective M4 mAChR antagonists may be effective therapeutics with fewer side effects than THP for the treatment of TOR1A dystonia.
1. Introduction
Dystonia associated with TOR1A variants, otherwise known as DYT1 dystonia, is an inherited form of dystonia caused by a three base pair deletion (Δgag) in the TOR1A gene (Ozelius et al., 1997). Because the precise mechanism(s) that lead to the abnormal movements are unknown, treatments for TOR1A dystonia are inadequate. The non-selective muscarinic acetylcholine receptor (mAChR) antagonist trihexyphenidyl (THP) is the preferred oral pharmaceutical for TOR1A dystonia, but its clinical use is limited due to significant dose-limiting side effects associated with its nonselective action at all five mAChRs subtypes (Schwarz and Bressman, 2009; Thenganatt and Jankovic, 2014). A better understanding of the mAChR subtype(s) responsible for the therapeutic effect of THP is critical for the development of targeted treatments with fewer side effects.
One potential mechanism of action of THP is the regulation of striatal dopamine (DA) neurotransmission. In patients, both DA metabolites and D2 DA receptor availability are abnormal (Augood et al., 2004; Asanuma et al., 2005). In mouse models, striatal DA release is reduced (Balcioglu et al., 2007; Page et al., 2010; Song et al., 2012; Downs et al., 2019). Striatal DA release is regulated by acetylcholine (ACh) (Zhang et al., 2002b; Zhang et al., 2002a; Exley and Cragg, 2008; Zhang et al., 2009a; Zhang et al., 2009b; Threlfell et al., 2010) and we recently discovered that THP rescues the deficit in both evoked DA release and steady-state extracellular DA concentrations in the striatum of a mouse knockin (KI) model of TOR1A dystonia (Tor1a+/ΔE KI mice) (Downs et al., 2019). However, the mAChR subtype(s) and striatal cell type that mediate the effects of THP are unknown.
THP binds to all 5 mAChRs with nanomolar affinity (Dorje et al., 1991; Bolden et al., 1992), and all 5 mAChRs are expressed in the striatum (Buckley et al., 1988; Levey et al., 1991; Harrison et al., 1996; Alcantara et al., 2001; Yamada et al., 2003; Hernandez-Flores et al., 2015). M1 mAChRs are expressed on both direct and indirect striatal projection neurons (dSPNs and iSPNs) where they mediate corticostriatal plasticity (Harrison et al., 1996; Hernandez-Flores et al., 2015). M1 mAChRs also regulate extracellular DA concentrations, although the underlying mechanisms are unknown (Gerber et al., 2001a). M4 mAChRs are expressed on dSPNs where they regulate corticostriatal plasticity (Nair et al., 2015; Nair et al., 2019) and mediate striatal DA release via endocannabinoid signaling (Foster et al., 2016). Additionally, M4 mAChRs expressed on cholinergic interneurons (ChIs) act as inhibitory autoreceptors to modulate extracellular ACh concentrations, which play a central role in mediating DA release (Threlfell et al., 2010; Pancani et al., 2014). While M2 and M3 mAChRs are located on glutamatergic axon terminals within the striatum (Buckley et al., 1988; Levey et al., 1991; Alcantara et al., 2001), we have previously demonstrated that THP does not depend on glutamatergic signaling to modulate striatal DA release (Downs et al., 2019). M5 mAChR are expressed exclusively on DA terminals in the striatum (Yamada et al., 2003). However, previous studies have demonstrated that activation of M5 mAChRs in the striatum stimulates DA release, so it is unlikely that the mAChR antagonist THP enhances DA release through this mechanism (Foster et al., 2014). Therefore, THP likely acts through M1 or M4 mAChRs or both to enhance DA release. In this study, we used a combination of pharmacologic challenges and cell-type specific mAChR knockout (KO) mice to determine the role of M1 and M4 mAChRs in the potentiation of DA release by THP in a KI mouse model of TOR1A dystonia (Tor1a+/ΔE).
2. Materials and methods
2.1. Animals
Male and female mice (8–14 weeks of age) inbred on C57BL/6 J were used for all studies. All mice were bred at Emory University. Animals were maintained on a 12 h light/dark cycle and allowed ad libitum access to food and water. All experimental procedures were approved by the Emory Animal Care and Use Committee and followed guidelines set forth in the Guide for the Care and Use of Laboratory Animals. Heterozygous knockin mice carrying the Tor1a(Δgag) mutation (Tor1a+/ΔE) (Goodchild et al., 2005) and normal littermates (Tor1a+/+) were genotyped using PCR (forward primer 5’-GCTATGGAAGCTCTAGTTGG-3′; reverse primer 5’-CAGCCAGGGCTAAACAGAG-3′). The following strains were used for the conditional mAChR KO mice experiments (Table 1): M4 mAChR flox (M4flox/flox) (Jeon et al., 2010), M1 mAChR flox (M1flox/flox) (Kamsler et al., 2010), ChAT-cre (ChATtm1(cre)/Lowl/MwarJ; JAX #031661), D1-cre (Tg(Drd1-cre)EY262Gsat/Mmucd; MMRRC #030989-UCD), and A2A-cre (Tg(Adora2a-cre)KG139Gsat/Mmucd; MMRRC #036158-UCD). These strains were used to generate both Tor1a+/+ and Tor1a+/ΔE mice with conditional KO of M4 mAChR from cholinergic neurons (ChAT-cre; M4flox/flox), KO of M4 mAChR from dSPNs (D1-cre; M4flox/flox), KO of M1 mAChR from dSPNs (D1-cre; M1flox/flox), and KO of M1 from iSPNs (A2A-cre; M1flox/flox). Mice were genotyped using PCR with the following primers D1-cre (forward primer 5′- GCTATGGAGATGCTCCTGATGGAA-3′; reverse primer 5′- CGGCAAACGGACAGAAGCATT -3′); A2A-cre (forward primer 5′- CGTGAGAAAGCCTTTGGGAAGCT-3′; reverse primer 5′- CGGCAAACGGACAGAAGCATT-3′); ChAT-cre (WT forward primer, 5’-GCAAAGAGACCTCATCTGTGGA-3′; cre forward primer, 5’-TTCACTGCATTCTAGTTGTGGT-3′; common reverse primer, 5’-GATAGGGGAGCAGCACACAG-3′); M1 flox (WT forward primer 5′- GAGCCTCAGTTTTCTCATTGG-3′; mutant forward primer 5′- AACACTACTTACACGTGGTGC-3′; common reverse primer 5′- TCAACCTGTACTGGTGATACG-3′); M4 flox (forward primer 5′- TGCAGATGTAGCTCAGCTCAGCGGTAC-3′; reverse primer 5′- TGAAGGTTGTAGACAAAGCTATACACATGGC-3′). Controls for the conditional KO experiments were homozygous for the flox'd muscarinic receptor allele but did not carry a cre transgene. All strains were maintained on a C57BL/6 J background.
Trihexyphenidyl (THP), a non-selective muscarinic receptor (mAChR) antagonist, is commonly used for the treatment of dystonia associated with TOR1A, otherwise known as DYT1 dystonia. A better understanding of the mechanism of action of THP is a critical step in the development of better therapeutics with fewer side effects. We previously found that THP normalizes the deficit in striatal dopamine (DA) release in a mouse model of TOR1A dystonia (Tor1a+/ΔE knockin (KI) mice), revealing a plausible mechanism of action for this compound, considering that abnormal DA neurotransmission is consistently associated with many forms of dystonia. However, the mAChR subtype(s) that mediate the rescue of striatal dopamine release remain unclear. In this study we used a combination of pharmacological challenges and cell-type specific mAChR conditional knockout mice of either sex to determine which mAChR subtypes mediate the DA release-enhancing effects of THP. We determined that THP acts in part at M4 mAChR on striatal cholinergic interneurons to enhance DA release in both Tor1a+/+ and Tor1a+/ΔE KI mice. Further, we found that the subtype selective M4 antagonist VU6021625 recapitulates the effects of THP. These data implicate a principal role for M4 mAChR located on striatal cholinergic interneurons in the mechanism of action of THP and suggest that subtype selective M4 mAChR antagonists may be effective therapeutics with fewer side effects than THP for the treatment of TOR1A dystonia.
1. Introduction
Dystonia associated with TOR1A variants, otherwise known as DYT1 dystonia, is an inherited form of dystonia caused by a three base pair deletion (Δgag) in the TOR1A gene (Ozelius et al., 1997). Because the precise mechanism(s) that lead to the abnormal movements are unknown, treatments for TOR1A dystonia are inadequate. The non-selective muscarinic acetylcholine receptor (mAChR) antagonist trihexyphenidyl (THP) is the preferred oral pharmaceutical for TOR1A dystonia, but its clinical use is limited due to significant dose-limiting side effects associated with its nonselective action at all five mAChRs subtypes (Schwarz and Bressman, 2009; Thenganatt and Jankovic, 2014). A better understanding of the mAChR subtype(s) responsible for the therapeutic effect of THP is critical for the development of targeted treatments with fewer side effects.
One potential mechanism of action of THP is the regulation of striatal dopamine (DA) neurotransmission. In patients, both DA metabolites and D2 DA receptor availability are abnormal (Augood et al., 2004; Asanuma et al., 2005). In mouse models, striatal DA release is reduced (Balcioglu et al., 2007; Page et al., 2010; Song et al., 2012; Downs et al., 2019). Striatal DA release is regulated by acetylcholine (ACh) (Zhang et al., 2002b; Zhang et al., 2002a; Exley and Cragg, 2008; Zhang et al., 2009a; Zhang et al., 2009b; Threlfell et al., 2010) and we recently discovered that THP rescues the deficit in both evoked DA release and steady-state extracellular DA concentrations in the striatum of a mouse knockin (KI) model of TOR1A dystonia (Tor1a+/ΔE KI mice) (Downs et al., 2019). However, the mAChR subtype(s) and striatal cell type that mediate the effects of THP are unknown.
THP binds to all 5 mAChRs with nanomolar affinity (Dorje et al., 1991; Bolden et al., 1992), and all 5 mAChRs are expressed in the striatum (Buckley et al., 1988; Levey et al., 1991; Harrison et al., 1996; Alcantara et al., 2001; Yamada et al., 2003; Hernandez-Flores et al., 2015). M1 mAChRs are expressed on both direct and indirect striatal projection neurons (dSPNs and iSPNs) where they mediate corticostriatal plasticity (Harrison et al., 1996; Hernandez-Flores et al., 2015). M1 mAChRs also regulate extracellular DA concentrations, although the underlying mechanisms are unknown (Gerber et al., 2001a). M4 mAChRs are expressed on dSPNs where they regulate corticostriatal plasticity (Nair et al., 2015; Nair et al., 2019) and mediate striatal DA release via endocannabinoid signaling (Foster et al., 2016). Additionally, M4 mAChRs expressed on cholinergic interneurons (ChIs) act as inhibitory autoreceptors to modulate extracellular ACh concentrations, which play a central role in mediating DA release (Threlfell et al., 2010; Pancani et al., 2014). While M2 and M3 mAChRs are located on glutamatergic axon terminals within the striatum (Buckley et al., 1988; Levey et al., 1991; Alcantara et al., 2001), we have previously demonstrated that THP does not depend on glutamatergic signaling to modulate striatal DA release (Downs et al., 2019). M5 mAChR are expressed exclusively on DA terminals in the striatum (Yamada et al., 2003). However, previous studies have demonstrated that activation of M5 mAChRs in the striatum stimulates DA release, so it is unlikely that the mAChR antagonist THP enhances DA release through this mechanism (Foster et al., 2014). Therefore, THP likely acts through M1 or M4 mAChRs or both to enhance DA release. In this study, we used a combination of pharmacologic challenges and cell-type specific mAChR knockout (KO) mice to determine the role of M1 and M4 mAChRs in the potentiation of DA release by THP in a KI mouse model of TOR1A dystonia (Tor1a+/ΔE).
2. Materials and methods
2.1. Animals
Male and female mice (8–14 weeks of age) inbred on C57BL/6 J were used for all studies. All mice were bred at Emory University. Animals were maintained on a 12 h light/dark cycle and allowed ad libitum access to food and water. All experimental procedures were approved by the Emory Animal Care and Use Committee and followed guidelines set forth in the Guide for the Care and Use of Laboratory Animals. Heterozygous knockin mice carrying the Tor1a(Δgag) mutation (Tor1a+/ΔE) (Goodchild et al., 2005) and normal littermates (Tor1a+/+) were genotyped using PCR (forward primer 5’-GCTATGGAAGCTCTAGTTGG-3′; reverse primer 5’-CAGCCAGGGCTAAACAGAG-3′). The following strains were used for the conditional mAChR KO mice experiments (Table 1): M4 mAChR flox (M4flox/flox) (Jeon et al., 2010), M1 mAChR flox (M1flox/flox) (Kamsler et al., 2010), ChAT-cre (ChATtm1(cre)/Lowl/MwarJ; JAX #031661), D1-cre (Tg(Drd1-cre)EY262Gsat/Mmucd; MMRRC #030989-UCD), and A2A-cre (Tg(Adora2a-cre)KG139Gsat/Mmucd; MMRRC #036158-UCD). These strains were used to generate both Tor1a+/+ and Tor1a+/ΔE mice with conditional KO of M4 mAChR from cholinergic neurons (ChAT-cre; M4flox/flox), KO of M4 mAChR from dSPNs (D1-cre; M4flox/flox), KO of M1 mAChR from dSPNs (D1-cre; M1flox/flox), and KO of M1 from iSPNs (A2A-cre; M1flox/flox). Mice were genotyped using PCR with the following primers D1-cre (forward primer 5′- GCTATGGAGATGCTCCTGATGGAA-3′; reverse primer 5′- CGGCAAACGGACAGAAGCATT -3′); A2A-cre (forward primer 5′- CGTGAGAAAGCCTTTGGGAAGCT-3′; reverse primer 5′- CGGCAAACGGACAGAAGCATT-3′); ChAT-cre (WT forward primer, 5’-GCAAAGAGACCTCATCTGTGGA-3′; cre forward primer, 5’-TTCACTGCATTCTAGTTGTGGT-3′; common reverse primer, 5’-GATAGGGGAGCAGCACACAG-3′); M1 flox (WT forward primer 5′- GAGCCTCAGTTTTCTCATTGG-3′; mutant forward primer 5′- AACACTACTTACACGTGGTGC-3′; common reverse primer 5′- TCAACCTGTACTGGTGATACG-3′); M4 flox (forward primer 5′- TGCAGATGTAGCTCAGCTCAGCGGTAC-3′; reverse primer 5′- TGAAGGTTGTAGACAAAGCTATACACATGGC-3′). Controls for the conditional KO experiments were homozygous for the flox'd muscarinic receptor allele but did not carry a cre transgene. All strains were maintained on a C57BL/6 J background.

2.2. Fluorescent in situ hybridization
Brains were rapidly dissected, frozen and kept at −80 °C until sectioning (10 μm). RNAscope fluorescent in situ hybridization was performed according to manufacturer's instructions (Advanced Cell Diagnostics, Newark, CA, USA). Briefly, slides were fixed in ice-cold 4% paraformaldehyde in 0.1 M PBS (phosphate buffered saline) for 15 mins. Slides were then dehydrated in graded ethanols and allowed to air dry for 5 min. Slides were treated with Protease-IV solution for 30 min at room temperature and then incubated with the appropriate probes for 2 h at 40 °C. The following probes were used: Mm-Chat-C1 (cat # 408731), Mm-Chrm4-C2 (cat # 410581-C2), Mm-Drd1-C3 (cat # 461901-C3), Mm-Chrm1-C1 (cat # 495291), Mm-Adora2a-C2 (cat # 409431-C2). A series of four amplification steps was then performed according to manufacturer's instructions with 2 × 2 min washes in wash buffer between each amplification step. Slides were counterstained with DAPI and then coverslipped using Prolong Glass Antifade Mounting media (Thermo Fisher Scientific, Waltham, MA, USA). Slides were stored at 4 °C until imaging.
Slides were imaged using a Leica SP8 confocal microscope using Leica Acquisition Suite (LAX S) (Leica Microsystems, Buffalo Grove, IL, USA). A total of 6 regions of interest (ROIs) from the dorsolateral quadrant of the striatum was taken from each section using a 40× or 60× objective and an optical thickness of ~5 μm. The same optical settings were used for image acquisition from each mouse with the settings titrated for each specific probe. Quantification of labeled cells was performed using the Cell Counter function in ImageJ (NIH). For each genotype, only cells that were labeled with RNAscope probes were counted, rather than counting all DAPI-positive cells in the striatum. Data were presented as both percentage of positively labeled cells for each marker as well as the number of cells for each probe. Cells were considered positive for a given probe if there were > 5 particles clustered around the nucleus.
2.3. Fast scan cyclic voltammetry
Fast scan cyclic voltammetry (FSCV) to measure DA release was performed according to previously published methods (Downs et al., 2019). Mice were euthanized using cervical dislocation and 300 μm sections were prepared using a vibratome in ice-cold oxygenated sucrose-supplemented artificial cerebral spinal fluid (aCSF) containing [in mM]: sucrose [194], NaCl [20], KCl [4.4], CaCl2 [1.2], MgCl2 [1.2], NaH2PO4 [1.2], NaHCO3 [25], d-glucose [11] at pH 7.4. After sectioning, brain slices were maintained in a holding chamber containing oxygenated bicarbonate-buffered aCSF containing [in mM]: NaCl [126], KCl [2.45], CaCl2 [2.4], MgCl2 [1.2], NaH2PO4 [1.2], NaHCO3 [25], d-glucose [11] and maintained at room temperature for 45–60 min before experiments began.
All FSCV experiments were performed in the dorsolateral striatum at ~ Bregma +0.26 mm because this region receives dense innervation from the motor cortex (Hintiryan et al., 2016). A slice was transferred to the recording chamber and perfused with oxygenated aCSF at 32 °C for 30 min to equilibrate to the chamber. A carbon fiber electrode constructed in-house was inserted approximately 50 μm into the surface of the slice and a bipolar tungsten stimulating electrode was placed approximately 200 μm away. DA release was evoked by a 1-pulse (600 μA, 4 ms pulse width) electrical stimulation at 5 min inter-stimulus intervals to prevent rundown. The scan rate for voltammetry was 400 V/s from −0.4 V to 1.3 V to −0.4 V verses Ag/AgCl with a sampling rate of 10 Hz using a Chem-Clamp voltammeter-amperometer (Dagan Corporation, Minneapolis, MN, USA). FSCV experiments were conducted and analyzed using Demon voltammetry software (Wake Forest University) (Yorgason et al., 2011) to determine peak DA release. All compounds were diluted in aCSF at the time of recording and bath applied for 10–20 min before recordings commenced to allow for equilibration. All electrodes were calibrated to known DA standards in aCSF using a custom-made flow cell.
2.4. Compounds
THP was purchased from Sigma-Aldrich (St. Louis, MO). Dihydro-β-erythroidine (DHβE) was obtained from Tocris (Minneapolis, MN). VU6021625 was generously provided by Dr. P. Jeffrey Conn (Vanderbilt University, TN).
2.5. Statistical analysis
All data are presented as means with standard error. For all FSCV experiments, we used one slice per animal, so all samples are independent. Dose response experiments were analyzed with non-linear regression to determine EC50. EC50 was analyzed using a two-tailed Student's t-test. Fluorescent in situ hybridization data was analyzed using one-way ANOVA with post hoc Dunnett's multiple comparison test. Experiments assessing DA release were analyzed using 2-way repeated measures ANOVA (genotype x treatment) with post hoc Sidak's multiple comparison test. Analyses were performed separately for Tor1a+/+ and Tor1a+/ΔE in mAChR KO experiments. All analyses were performed using Graphpad Prism 9 (https://www.graphpad.com/). Statistical significance is defined as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3. Results
3.1. M4 mAChRs in striatal cholinergic cells mediate the effects of THP
We have previously demonstrated that the non-selective mAChR antagonist THP enhances DA release in both Tor1a+/+ and Tor1a+/ΔE KI mice and rescues the deficiency in DA release in Tor1a+/ΔE KI mice (Downs et al., 2019). However, the mAChR subtype(s) and cell types that mediate this effect are unknown. M4 mAChRs expressed on dSPNs, ChIs, and cortical glutamatergic terminals are known to regulate striatal DA release (Harrison et al., 1996; Threlfell et al., 2010; Threlfell and Cragg, 2011; Pancani et al., 2014). Therefore, to determine if THP depends on M4 mAChRs on any of these cell types, we assessed striatal DA release in response to challenge with THP in Tor1a+/+ and Tor1a+/ΔE KI mice lacking M4 mAChRs on ChIs (ChI-M4−/−) or M4 mAChRs on dSPNs (dSPN-M4−/−). Deletion of M4 mAChRs from cortical glutamatergic inputs was not investigated because we have previously demonstrated that blocking ionotropic glutamate receptors does not affect the ability of THP to enhance DA release (Downs et al., 2019).
Previous studies have demonstrated that M4 mAChRs on dSPNs regulate striatal DA release by modulating endocannabinoid production (Foster et al., 2016). To test the hypothesis that THP enhances DA release by acting on dSPNs, we measured electrically evoked DA release in striatal slices from mice lacking M4 mAChRs in direct SPNs (dSPN-M4−/−) in the presence of THP (300 nM). Male and female Tor1a+/+ mice had a similar response to THP, so male and female mice were combined for all studies (main effect of sex, F1,8 = 0.039, p = 0.85, treatment x sex interaction effect, F1,8 = 0.00012, p = 0.99). THP did not alter DA clearance (tau), which is a surrogate for reuptake, in either Tor1a+/+ or Tor1a+/ΔE mice (main effect of treatment, F1,6 = 0.046, p = 0.84). The dSPN-M4 KO had no effect on baseline DA release in either Tor1a+/+ mice (Sidak's multiple comparison test, p = 0.78) or Tor1a+/ΔE (Sidak's multiple comparison test, p = 0.99). Deleting M4 mAChR expression from dSPNs had no effect on the THP-induced increase in DA release in either Tor1a+/+ mice (main effect of treatment, F2,12 = 144.3, p < 0.0001; Sidak's multiple comparisons test, p < 0.0001) or Tor1a+/ΔE mice (Fig. 1A, main effect of treatment, F2,12 = 71.71, p < 0.0001; Sidak's multiple comparison test, p < 0.0001).
Brains were rapidly dissected, frozen and kept at −80 °C until sectioning (10 μm). RNAscope fluorescent in situ hybridization was performed according to manufacturer's instructions (Advanced Cell Diagnostics, Newark, CA, USA). Briefly, slides were fixed in ice-cold 4% paraformaldehyde in 0.1 M PBS (phosphate buffered saline) for 15 mins. Slides were then dehydrated in graded ethanols and allowed to air dry for 5 min. Slides were treated with Protease-IV solution for 30 min at room temperature and then incubated with the appropriate probes for 2 h at 40 °C. The following probes were used: Mm-Chat-C1 (cat # 408731), Mm-Chrm4-C2 (cat # 410581-C2), Mm-Drd1-C3 (cat # 461901-C3), Mm-Chrm1-C1 (cat # 495291), Mm-Adora2a-C2 (cat # 409431-C2). A series of four amplification steps was then performed according to manufacturer's instructions with 2 × 2 min washes in wash buffer between each amplification step. Slides were counterstained with DAPI and then coverslipped using Prolong Glass Antifade Mounting media (Thermo Fisher Scientific, Waltham, MA, USA). Slides were stored at 4 °C until imaging.
Slides were imaged using a Leica SP8 confocal microscope using Leica Acquisition Suite (LAX S) (Leica Microsystems, Buffalo Grove, IL, USA). A total of 6 regions of interest (ROIs) from the dorsolateral quadrant of the striatum was taken from each section using a 40× or 60× objective and an optical thickness of ~5 μm. The same optical settings were used for image acquisition from each mouse with the settings titrated for each specific probe. Quantification of labeled cells was performed using the Cell Counter function in ImageJ (NIH). For each genotype, only cells that were labeled with RNAscope probes were counted, rather than counting all DAPI-positive cells in the striatum. Data were presented as both percentage of positively labeled cells for each marker as well as the number of cells for each probe. Cells were considered positive for a given probe if there were > 5 particles clustered around the nucleus.
2.3. Fast scan cyclic voltammetry
Fast scan cyclic voltammetry (FSCV) to measure DA release was performed according to previously published methods (Downs et al., 2019). Mice were euthanized using cervical dislocation and 300 μm sections were prepared using a vibratome in ice-cold oxygenated sucrose-supplemented artificial cerebral spinal fluid (aCSF) containing [in mM]: sucrose [194], NaCl [20], KCl [4.4], CaCl2 [1.2], MgCl2 [1.2], NaH2PO4 [1.2], NaHCO3 [25], d-glucose [11] at pH 7.4. After sectioning, brain slices were maintained in a holding chamber containing oxygenated bicarbonate-buffered aCSF containing [in mM]: NaCl [126], KCl [2.45], CaCl2 [2.4], MgCl2 [1.2], NaH2PO4 [1.2], NaHCO3 [25], d-glucose [11] and maintained at room temperature for 45–60 min before experiments began.
All FSCV experiments were performed in the dorsolateral striatum at ~ Bregma +0.26 mm because this region receives dense innervation from the motor cortex (Hintiryan et al., 2016). A slice was transferred to the recording chamber and perfused with oxygenated aCSF at 32 °C for 30 min to equilibrate to the chamber. A carbon fiber electrode constructed in-house was inserted approximately 50 μm into the surface of the slice and a bipolar tungsten stimulating electrode was placed approximately 200 μm away. DA release was evoked by a 1-pulse (600 μA, 4 ms pulse width) electrical stimulation at 5 min inter-stimulus intervals to prevent rundown. The scan rate for voltammetry was 400 V/s from −0.4 V to 1.3 V to −0.4 V verses Ag/AgCl with a sampling rate of 10 Hz using a Chem-Clamp voltammeter-amperometer (Dagan Corporation, Minneapolis, MN, USA). FSCV experiments were conducted and analyzed using Demon voltammetry software (Wake Forest University) (Yorgason et al., 2011) to determine peak DA release. All compounds were diluted in aCSF at the time of recording and bath applied for 10–20 min before recordings commenced to allow for equilibration. All electrodes were calibrated to known DA standards in aCSF using a custom-made flow cell.
2.4. Compounds
THP was purchased from Sigma-Aldrich (St. Louis, MO). Dihydro-β-erythroidine (DHβE) was obtained from Tocris (Minneapolis, MN). VU6021625 was generously provided by Dr. P. Jeffrey Conn (Vanderbilt University, TN).
2.5. Statistical analysis
All data are presented as means with standard error. For all FSCV experiments, we used one slice per animal, so all samples are independent. Dose response experiments were analyzed with non-linear regression to determine EC50. EC50 was analyzed using a two-tailed Student's t-test. Fluorescent in situ hybridization data was analyzed using one-way ANOVA with post hoc Dunnett's multiple comparison test. Experiments assessing DA release were analyzed using 2-way repeated measures ANOVA (genotype x treatment) with post hoc Sidak's multiple comparison test. Analyses were performed separately for Tor1a+/+ and Tor1a+/ΔE in mAChR KO experiments. All analyses were performed using Graphpad Prism 9 (https://www.graphpad.com/). Statistical significance is defined as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3. Results
3.1. M4 mAChRs in striatal cholinergic cells mediate the effects of THP
We have previously demonstrated that the non-selective mAChR antagonist THP enhances DA release in both Tor1a+/+ and Tor1a+/ΔE KI mice and rescues the deficiency in DA release in Tor1a+/ΔE KI mice (Downs et al., 2019). However, the mAChR subtype(s) and cell types that mediate this effect are unknown. M4 mAChRs expressed on dSPNs, ChIs, and cortical glutamatergic terminals are known to regulate striatal DA release (Harrison et al., 1996; Threlfell et al., 2010; Threlfell and Cragg, 2011; Pancani et al., 2014). Therefore, to determine if THP depends on M4 mAChRs on any of these cell types, we assessed striatal DA release in response to challenge with THP in Tor1a+/+ and Tor1a+/ΔE KI mice lacking M4 mAChRs on ChIs (ChI-M4−/−) or M4 mAChRs on dSPNs (dSPN-M4−/−). Deletion of M4 mAChRs from cortical glutamatergic inputs was not investigated because we have previously demonstrated that blocking ionotropic glutamate receptors does not affect the ability of THP to enhance DA release (Downs et al., 2019).
Previous studies have demonstrated that M4 mAChRs on dSPNs regulate striatal DA release by modulating endocannabinoid production (Foster et al., 2016). To test the hypothesis that THP enhances DA release by acting on dSPNs, we measured electrically evoked DA release in striatal slices from mice lacking M4 mAChRs in direct SPNs (dSPN-M4−/−) in the presence of THP (300 nM). Male and female Tor1a+/+ mice had a similar response to THP, so male and female mice were combined for all studies (main effect of sex, F1,8 = 0.039, p = 0.85, treatment x sex interaction effect, F1,8 = 0.00012, p = 0.99). THP did not alter DA clearance (tau), which is a surrogate for reuptake, in either Tor1a+/+ or Tor1a+/ΔE mice (main effect of treatment, F1,6 = 0.046, p = 0.84). The dSPN-M4 KO had no effect on baseline DA release in either Tor1a+/+ mice (Sidak's multiple comparison test, p = 0.78) or Tor1a+/ΔE (Sidak's multiple comparison test, p = 0.99). Deleting M4 mAChR expression from dSPNs had no effect on the THP-induced increase in DA release in either Tor1a+/+ mice (main effect of treatment, F2,12 = 144.3, p < 0.0001; Sidak's multiple comparisons test, p < 0.0001) or Tor1a+/ΔE mice (Fig. 1A, main effect of treatment, F2,12 = 71.71, p < 0.0001; Sidak's multiple comparison test, p < 0.0001).
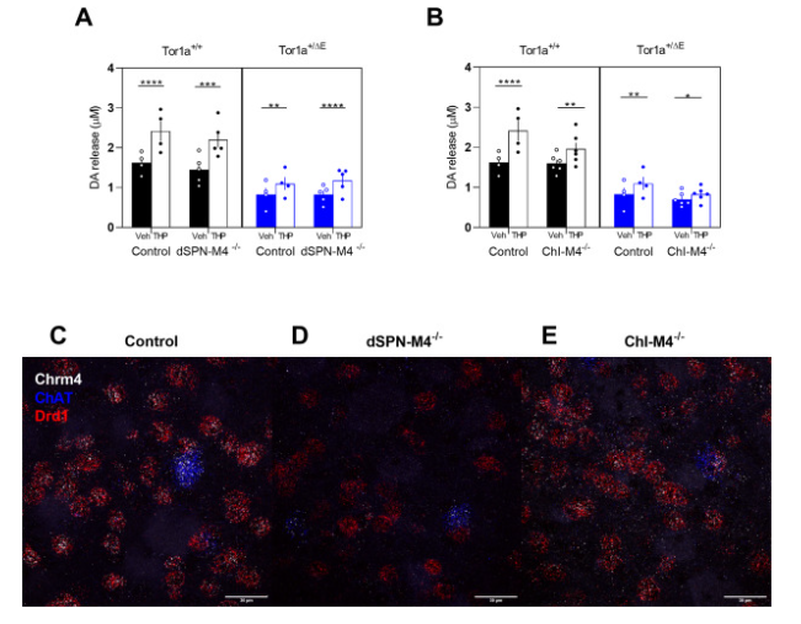
Fig. 1. Effect of cell-type specific deletion of M4 mAChRs on striatal DA release in Tor1a+/+ and Tor1a+/ΔE KI mice. A. KO of M4 mAChR from direct SPNs did not significantly change the DA-release enhancing effects of 300 nM THP in either genotype. B. KO of M4 mAChR from ChIs reduced the effect of THP compared to control mice that were homozygous for the flox'd M4 mAChR allele but did not carry a cre transgene. Data are expressed as mean peak concentration of DA released (μM) ± SEM (n = 4–6); *p < 0.05, **p < 0.01, ****p < 0.0001. C, D, E. Representative images of fluorescent in situ hybridization using probes to ChAT, Chrm4, and Drd1 mRNA in control C., dSPN-M4−/− D., and ChI-M4−/− E. mice demonstrate cell-type specific deletion of M4 mAChR. Scale bar = 30 μm.
To verify that the M4 mAChR conditional knockout in dSPNs was both effective and cell-type specific, we used fluorescent in situ hybridization with probes to Chrm4, Drd1 and ChAT to assess striatal mRNA expression. Chrm4 encodes the M4 mAChR. Drd1 encodes the D1 DA receptor which is a marker for dSPNs and ChAT encodes choline acetyltransferase, a marker for ChIs. In control mice, Chrm4 mRNA was expressed in all ChAT-positive cells and almost all Drd1-positive cells, as expected. In dSPN-M4−/− mice, there was a significant reduction in the percent of cells expressing both Chrm4 and Drd1 mRNA to ~13% of control mice while there was a corresponding increase in the percent of cells that expressed only Drd1 without Chrm4 (Fig. 1C vs D & Table 2, one-way ANVOA, F2,6 = 554.7, p < 0.0001). There was no change in Chrm4 mRNA expression in ChAT-positive striatal ChIs in dSPN-M4−/− compared to control mice. These results validate the cell-type specific conditional knockout of M4 mAChRs in dSPNs but not ChIs.
To verify that the M4 mAChR conditional knockout in dSPNs was both effective and cell-type specific, we used fluorescent in situ hybridization with probes to Chrm4, Drd1 and ChAT to assess striatal mRNA expression. Chrm4 encodes the M4 mAChR. Drd1 encodes the D1 DA receptor which is a marker for dSPNs and ChAT encodes choline acetyltransferase, a marker for ChIs. In control mice, Chrm4 mRNA was expressed in all ChAT-positive cells and almost all Drd1-positive cells, as expected. In dSPN-M4−/− mice, there was a significant reduction in the percent of cells expressing both Chrm4 and Drd1 mRNA to ~13% of control mice while there was a corresponding increase in the percent of cells that expressed only Drd1 without Chrm4 (Fig. 1C vs D & Table 2, one-way ANVOA, F2,6 = 554.7, p < 0.0001). There was no change in Chrm4 mRNA expression in ChAT-positive striatal ChIs in dSPN-M4−/− compared to control mice. These results validate the cell-type specific conditional knockout of M4 mAChRs in dSPNs but not ChIs.
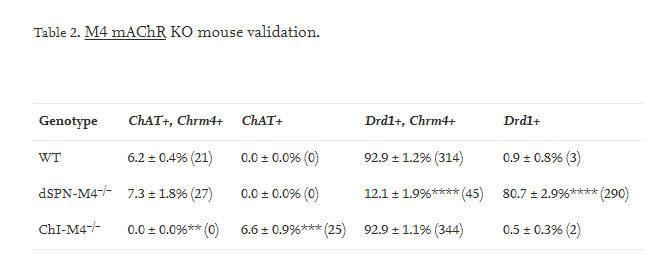
Data represent mean percentage ± SEM of cells positively labeled using in situ hybridization probes for ChAT, Chrm4, or Drd1 with the number of cells in parentheses. Only cells that express either ChAT, Chrm4, or Drd1 were included in the count. Data within each column were analyzed with one-way ANOVA with post hoc Dunnett's multiple comparisons test. **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to control.
M4 mAChRs are expressed on ChIs where they act as inhibitory autoreceptors. It is known that activation of these autoreceptors results in a reduction in DA release via a decrease in nicotinic acetylcholine receptor (nAChR) activation on DA terminals (Threlfell et al., 2010). To test the hypothesis that THP exerts its effect by blocking M4 mAChRs expressed on ChIs, we assessed DA release in striatal slices from mice lacking M4 mAChRs in ChIs (ChI-M4−/−). KO of M4 mAChRs from ChIs had no effect on baseline DA release in either Tor1a+/+ (Sidak's multiple comparison test, p = 0.99) or Tor1a+/ΔE mice (Sidak's multiple comparison test, p = 0.32). While THP increased DA release in both Tor1a+/+ and Tor1a+/+; ChI-M4−/− mice (Fig. 1B, main effect of treatment F2,12 = 144.3, p < 0.0001), the effect of THP was significantly attenuated in Tor1a+/+; ChI-M4−/− mice to 65% of Tor1a+/+ mice (treatment x genotype interaction effect, F2,12 = 7.43, p = 0.0079, Sidak's multiple comparison test, p = 0.0027). In Tor1a+/ΔE mice, THP significantly increased DA release (main effect of treatment, F1,12 = 71.71, p < 0.0001; treatment x genotype interaction effect F2,12 = 7.31, p = 0.0084). While THP increased DA release in Tor1a+/ΔE; ChI-M4−/− mice (Sidak's multiple comparisons test, p = 0.04), the effects of THP were attenuated from a 32% increase in DA release in control animals, to a 17% increase in Tor1a+/ΔE; ChI-M4−/− mice. However, this decrease did not reach statistical significance (Sidak's multiple comparisons test, p = 0.13). These results suggest that M4 mAChRs on ChIs mediate the effects of THP, in part, in both Tor1a+/+ and Tor1a+/ΔE KI mice.
Fluorescent in situ hybridization with probes to Chrm4, Drd1 and ChAT was used to verify that the M4 mAChR conditional knockout in ChIs was accomplished. In control mice, Chrm4 mRNA was detected in all ChAT-positive cells. However, in ChI-M4−/− mice, Chrm4 mRNA expression was not detected in any ChAT-positive cells (Fig. 1C vs E & Table 2, one-way ANVOA F2,6 = 18.68, p = 0.0002). There was no change in Chrm4 mRNA expression in Drd1-positive cells in ChI-M4−/− compared to control mice. Thus, the conditional Chrm4 deletion targeted to ChIs was cell-type specific and highly effective.
3.2. A selective M4 mAChR antagonist is sufficient to recapitulate the effects of THP
The cell-type selective KO experiments suggest that M4 mAChRs are necessary for the DA release-enhancing effects of THP in Tor1a+/ΔE KI mice. However, because THP acts at all five mAChRs, it is not clear if selective blockade of M4 mAChRs would rescue DA release in Tor1a+/ΔE KI mice similar to the effects of THP. Therefore, we assessed DA release in the presence of the subtype-selective M4 mAChR antagonist VU6021625, which was shown to be effective in ameliorating the dystonia exhibited by a mouse model of DOPA-response dystonia (Moehle et al., 2020). VU6021625 dose-dependently enhanced DA release in both Tor1a+/+ and Tor1a+/ΔE KI mice (Fig. 2A). There was no significant difference in EC50 between genotypes (Student's t-test, p = 0.87). VU6021625 did not alter DA clearance kinetics in either genotype (main effect of treatment, F1,6 = 0.002, p = 0.96. DA release was significantly increased in both Tor1a+/+ and Tor1a+/ΔE KI mice at the maximal concentration of 300 nM VU6021625 (Fig. 2B, main effect of treatment, F1,8 = 30.19, p = 0.0006). VU6021625 was similarly effective in both Tor1a+/+ and Tor1a+/ΔE KI mice (genotype x treatment interaction effect, F1,8 = 1.37, p = 0.27). Importantly, VU6021625 normalized DA release in Tor1a+/ΔE KI mice to Tor1a+/+ levels (Sidak's multiple comparison's test, p = 0.58).
M4 mAChRs are expressed on ChIs where they act as inhibitory autoreceptors. It is known that activation of these autoreceptors results in a reduction in DA release via a decrease in nicotinic acetylcholine receptor (nAChR) activation on DA terminals (Threlfell et al., 2010). To test the hypothesis that THP exerts its effect by blocking M4 mAChRs expressed on ChIs, we assessed DA release in striatal slices from mice lacking M4 mAChRs in ChIs (ChI-M4−/−). KO of M4 mAChRs from ChIs had no effect on baseline DA release in either Tor1a+/+ (Sidak's multiple comparison test, p = 0.99) or Tor1a+/ΔE mice (Sidak's multiple comparison test, p = 0.32). While THP increased DA release in both Tor1a+/+ and Tor1a+/+; ChI-M4−/− mice (Fig. 1B, main effect of treatment F2,12 = 144.3, p < 0.0001), the effect of THP was significantly attenuated in Tor1a+/+; ChI-M4−/− mice to 65% of Tor1a+/+ mice (treatment x genotype interaction effect, F2,12 = 7.43, p = 0.0079, Sidak's multiple comparison test, p = 0.0027). In Tor1a+/ΔE mice, THP significantly increased DA release (main effect of treatment, F1,12 = 71.71, p < 0.0001; treatment x genotype interaction effect F2,12 = 7.31, p = 0.0084). While THP increased DA release in Tor1a+/ΔE; ChI-M4−/− mice (Sidak's multiple comparisons test, p = 0.04), the effects of THP were attenuated from a 32% increase in DA release in control animals, to a 17% increase in Tor1a+/ΔE; ChI-M4−/− mice. However, this decrease did not reach statistical significance (Sidak's multiple comparisons test, p = 0.13). These results suggest that M4 mAChRs on ChIs mediate the effects of THP, in part, in both Tor1a+/+ and Tor1a+/ΔE KI mice.
Fluorescent in situ hybridization with probes to Chrm4, Drd1 and ChAT was used to verify that the M4 mAChR conditional knockout in ChIs was accomplished. In control mice, Chrm4 mRNA was detected in all ChAT-positive cells. However, in ChI-M4−/− mice, Chrm4 mRNA expression was not detected in any ChAT-positive cells (Fig. 1C vs E & Table 2, one-way ANVOA F2,6 = 18.68, p = 0.0002). There was no change in Chrm4 mRNA expression in Drd1-positive cells in ChI-M4−/− compared to control mice. Thus, the conditional Chrm4 deletion targeted to ChIs was cell-type specific and highly effective.
3.2. A selective M4 mAChR antagonist is sufficient to recapitulate the effects of THP
The cell-type selective KO experiments suggest that M4 mAChRs are necessary for the DA release-enhancing effects of THP in Tor1a+/ΔE KI mice. However, because THP acts at all five mAChRs, it is not clear if selective blockade of M4 mAChRs would rescue DA release in Tor1a+/ΔE KI mice similar to the effects of THP. Therefore, we assessed DA release in the presence of the subtype-selective M4 mAChR antagonist VU6021625, which was shown to be effective in ameliorating the dystonia exhibited by a mouse model of DOPA-response dystonia (Moehle et al., 2020). VU6021625 dose-dependently enhanced DA release in both Tor1a+/+ and Tor1a+/ΔE KI mice (Fig. 2A). There was no significant difference in EC50 between genotypes (Student's t-test, p = 0.87). VU6021625 did not alter DA clearance kinetics in either genotype (main effect of treatment, F1,6 = 0.002, p = 0.96. DA release was significantly increased in both Tor1a+/+ and Tor1a+/ΔE KI mice at the maximal concentration of 300 nM VU6021625 (Fig. 2B, main effect of treatment, F1,8 = 30.19, p = 0.0006). VU6021625 was similarly effective in both Tor1a+/+ and Tor1a+/ΔE KI mice (genotype x treatment interaction effect, F1,8 = 1.37, p = 0.27). Importantly, VU6021625 normalized DA release in Tor1a+/ΔE KI mice to Tor1a+/+ levels (Sidak's multiple comparison's test, p = 0.58).
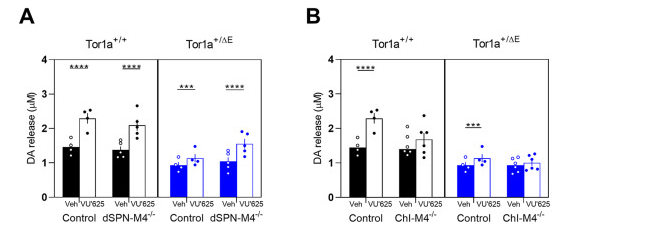
Fig. 3. Effect of M4 mAChR KO in dSPNs or ChIs on the VU6021625-induced increase in DA release A. KO of M4 mAChR from dSPNs did not significantly affect the DA release-enhancing effects of 300 nM VU6021625 compared to control mice that were homozygous for the flox'd M4 mAChR allele but did not carry a cre transgene. B. KO of M4 mAChR from ChIs reduced the DA release enhancing effects of VU6021625 in both Tor1a+/+ and Tor1a+/ΔE mice. Data are expressed as concentration of DA released (μM) ± SEM (n = 4–6); **p < 0.01, ****p < 0.0001.
Next, we determined the role of M4 mAChR expressed on ChIs in mediating the increase in DA release in response to VU6021625 treatment. KO of M4 mAChR from ChIs eliminated the DA enhancing effect of VU6021625 in Tor1a+/+ mice (Fig. 3B, treatment x genotype interaction effect, F212 = 15.84, p = 0.0004, Sidak's multiple comparison test, p = 0.27). KO of M4 mAChR from cholinergic neurons also eliminated the DA enhancing effect of VU6021625 in Tor1a+/ΔE KI mice (treatment x genotype interaction effect, F2,12 = 40.10, p < 0.0001, Sidak's multiple comparison test, p = 0.13). These data suggest that the M4 subtype selective mAChR antagonist enhances DA release by blocking M4 mAChR on ChIs in both Tor1a+/+ and Tor1a+/ΔE KI mice.
3.4. M1 mAChRs on dSPNs and iSPNs are not required for the effects of THP in Tor1a+/+ or Tor1a+/ΔE KI mice
M1 mAChRs are expressed on both direct and indirect SPNs (Harrison et al., 1996; Hernandez-Flores et al., 2015). Previous studies have demonstrated that M1 mAChR regulates extracellular DA in mice (Gerber et al., 2001a). Although the mechanism mediating this effect is largely unknown, it is known that SPNs provide GABAergic input to ChIs (Tepper et al., 2008). It is possible that THP mediates DA release indirectly by blocking excitatory M1 mAChRs on SPNs to disinhibit ChI activity thereby increasing ACh release and nAChR activity on DA terminals. To determine if M1 mAChRs on either direct or indirect SPNs are required for the DA release-enhancing effects of THP, we assessed striatal DA release in response to challenge with THP in Tor1a+/+ and Tor1a+/ΔE KI mice lacking M1 mAChR on dSPNs (dSPN-M1−/−) or iSPNs (iSPN-M1−/−).
In Tor1a+/+ mice lacking M1 mAChR in dSPNs, THP significantly increased DA release in both Tor1a+/+ and Tor1a+/+ dSPN-M1−/− mice (Fig. 4A treatment effect, F1,13 = 100.3, p < 0.0001, Sidak multiple comparisons test, p = 0.0007). Although there was a trend toward reduced efficacy in Tor1a+/+ dSPN-M1−/− mice (treatment x genotype interaction effect, F2,13 = 3.30, p = 0.069), KO of M1 mAChR from dSPNs did not affect the response to THP in Tor1a+/ΔE KI mice (treatment x genotype interaction effect F2,13 = 0.37, p = 0.72). There were no differences observed in baseline DA release in either Tor1a+/+ (Sidak's multiple comparison test, p = 0.98) or Tor1a+/ΔE (Sidak's multiple comparison test, p = 0.57) due to dSPN-M1 KO. These results demonstrate that M1 mAChRs on dSPNs do not significantly contribute to the effects of THP on DA release in either Tor1a+/+ or Tor1a+/ΔE KI mice.
Next, we determined the role of M4 mAChR expressed on ChIs in mediating the increase in DA release in response to VU6021625 treatment. KO of M4 mAChR from ChIs eliminated the DA enhancing effect of VU6021625 in Tor1a+/+ mice (Fig. 3B, treatment x genotype interaction effect, F212 = 15.84, p = 0.0004, Sidak's multiple comparison test, p = 0.27). KO of M4 mAChR from cholinergic neurons also eliminated the DA enhancing effect of VU6021625 in Tor1a+/ΔE KI mice (treatment x genotype interaction effect, F2,12 = 40.10, p < 0.0001, Sidak's multiple comparison test, p = 0.13). These data suggest that the M4 subtype selective mAChR antagonist enhances DA release by blocking M4 mAChR on ChIs in both Tor1a+/+ and Tor1a+/ΔE KI mice.
3.4. M1 mAChRs on dSPNs and iSPNs are not required for the effects of THP in Tor1a+/+ or Tor1a+/ΔE KI mice
M1 mAChRs are expressed on both direct and indirect SPNs (Harrison et al., 1996; Hernandez-Flores et al., 2015). Previous studies have demonstrated that M1 mAChR regulates extracellular DA in mice (Gerber et al., 2001a). Although the mechanism mediating this effect is largely unknown, it is known that SPNs provide GABAergic input to ChIs (Tepper et al., 2008). It is possible that THP mediates DA release indirectly by blocking excitatory M1 mAChRs on SPNs to disinhibit ChI activity thereby increasing ACh release and nAChR activity on DA terminals. To determine if M1 mAChRs on either direct or indirect SPNs are required for the DA release-enhancing effects of THP, we assessed striatal DA release in response to challenge with THP in Tor1a+/+ and Tor1a+/ΔE KI mice lacking M1 mAChR on dSPNs (dSPN-M1−/−) or iSPNs (iSPN-M1−/−).
In Tor1a+/+ mice lacking M1 mAChR in dSPNs, THP significantly increased DA release in both Tor1a+/+ and Tor1a+/+ dSPN-M1−/− mice (Fig. 4A treatment effect, F1,13 = 100.3, p < 0.0001, Sidak multiple comparisons test, p = 0.0007). Although there was a trend toward reduced efficacy in Tor1a+/+ dSPN-M1−/− mice (treatment x genotype interaction effect, F2,13 = 3.30, p = 0.069), KO of M1 mAChR from dSPNs did not affect the response to THP in Tor1a+/ΔE KI mice (treatment x genotype interaction effect F2,13 = 0.37, p = 0.72). There were no differences observed in baseline DA release in either Tor1a+/+ (Sidak's multiple comparison test, p = 0.98) or Tor1a+/ΔE (Sidak's multiple comparison test, p = 0.57) due to dSPN-M1 KO. These results demonstrate that M1 mAChRs on dSPNs do not significantly contribute to the effects of THP on DA release in either Tor1a+/+ or Tor1a+/ΔE KI mice.
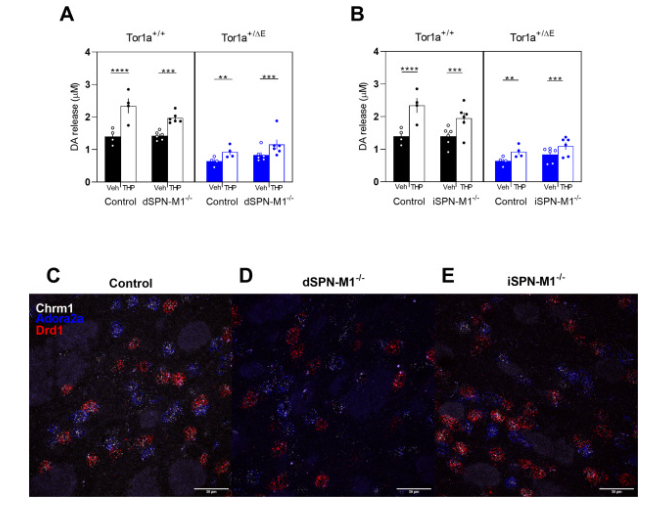
Fig. 4. Effect of cell-type specific deletion of M1 mAChRs on striatal DA release in Tor1a+/+ and Tor1a+/ΔE KI mice A. KO of M1 mAChR from direct SPNs did not alter the effects of THP in Tor1a+/+ or Tor1a+/ΔE KI mice compared to control mice that were homozygous for the flox'd M1 mAChR allele but did not carry a cre transgene. B. KO of M1 mAChR from indirect SPNs did not alter the effects of THP in Tor1a+/+ or Tor1a+/ΔE KI mice compared to control mice that were homozygous for the flox'd M1 mAChR allele but did not carry a cre transgene. Data are expressed as concentration of DA released (μM) ± SEM. (n = 4–6); *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Representative images of control C., dSPN-M4−/− D., and iSPN-M4−/− E. mice labeled for ChAT, Chrm4, and Drd1 mRNA. Scale bar = 30 μm.
In Tor1a+/+ mice lacking M1 mAChR in iSPNs, THP significantly increased DA release (Fig. 4B, main effect of treatment, F1,13 = 100.3, p < 0.0001). However, there was a trend toward reduced efficacy in Tor1a+/+ iSPN-M1−/− mice (treatment x genotype interaction effect, F2,13 = 3.04, p = 0.069). The effect of THP in Tor1a+/ΔE iSPN-M1−/− mice did not differ from Tor1a+/ΔE mice (treatment x genotype interaction effect F2,13 = 0.34, p = 0.72). Deletion of M1 mAChRs from iSPNs had no effect on baseline release in either Tor1a+/+ (Sidak's multiple comparison test, p < 0.99) or Tor1a+/ΔE (Sidak's multiple comparison test, p < 0.43). These results demonstrate that M1 mAChRs on iSPNs do not significantly contribute to the effects of THP on DA release in either Tor1a+/+ or Tor1a+/ΔE KI mice.
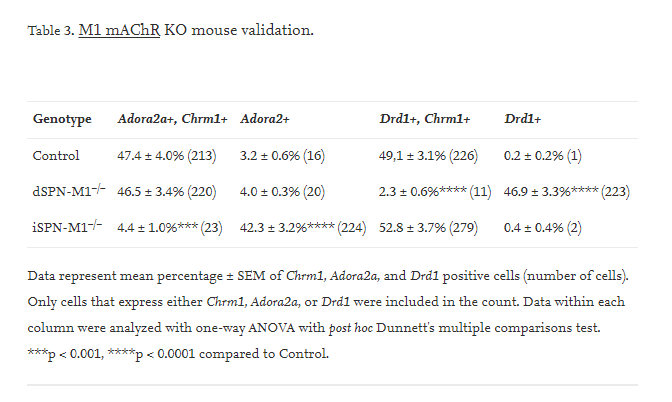
To confirm that the SPN cell-type specific KO of M1 mAChRs were effective, we used fluorescent in situ hybridization with probes specific to Chrm1, Adora2a, and Drd1. Chrm1 encodes the M1 mAChR receptor. Adora2a encodes the A2A adenosine receptor and is a specific marker for iSPNs. Drd1 is a specific marker for dSPNs. In control mice, Chrm1 was expressed in almost all Adora2a-positive and Drd1-positive cells. However, in dSPN-M1−/− mice, the percentage of cells expressing both Chrm1 and Drd1 mRNA was significantly reduced to ~2% of control levels (Fig. 4C vs D & Table 3, one-way ANVOA F2,6 = 90.44, p < 0.0001) while the percentage of cells expressing both Chrm1 and Adora2a was unchanged. In iSPN-M1−/−mice, the percentage of cells expressing both Chrm1 and Adora2a mRNA was significantly reduced to ~5% of control levels (Fig. 4C vs E & Table 3, one-way ANOVA F2,6 = 57.37, p < 0.0001), while the percentage of cells expressing both Chrm1 and Drd1 mRNA was unchanged. These results demonstrate that the M1 mAChR knockouts were specific and effective.
In Tor1a+/+ mice lacking M1 mAChR in iSPNs, THP significantly increased DA release (Fig. 4B, main effect of treatment, F1,13 = 100.3, p < 0.0001). However, there was a trend toward reduced efficacy in Tor1a+/+ iSPN-M1−/− mice (treatment x genotype interaction effect, F2,13 = 3.04, p = 0.069). The effect of THP in Tor1a+/ΔE iSPN-M1−/− mice did not differ from Tor1a+/ΔE mice (treatment x genotype interaction effect F2,13 = 0.34, p = 0.72). Deletion of M1 mAChRs from iSPNs had no effect on baseline release in either Tor1a+/+ (Sidak's multiple comparison test, p < 0.99) or Tor1a+/ΔE (Sidak's multiple comparison test, p < 0.43). These results demonstrate that M1 mAChRs on iSPNs do not significantly contribute to the effects of THP on DA release in either Tor1a+/+ or Tor1a+/ΔE KI mice.
To confirm that the SPN cell-type specific KO of M1 mAChRs were effective, we used fluorescent in situ hybridization with probes specific to Chrm1, Adora2a, and Drd1. Chrm1 encodes the M1 mAChR receptor. Adora2a encodes the A2A adenosine receptor and is a specific marker for iSPNs. Drd1 is a specific marker for dSPNs. In control mice, Chrm1 was expressed in almost all Adora2a-positive and Drd1-positive cells. However, in dSPN-M1−/− mice, the percentage of cells expressing both Chrm1 and Drd1 mRNA was significantly reduced to ~2% of control levels (Fig. 4C vs D & Table 3, one-way ANVOA F2,6 = 90.44, p < 0.0001) while the percentage of cells expressing both Chrm1 and Adora2a was unchanged. In iSPN-M1−/−mice, the percentage of cells expressing both Chrm1 and Adora2a mRNA was significantly reduced to ~5% of control levels (Fig. 4C vs E & Table 3, one-way ANOVA F2,6 = 57.37, p < 0.0001), while the percentage of cells expressing both Chrm1 and Drd1 mRNA was unchanged. These results demonstrate that the M1 mAChR knockouts were specific and effective.
4. Discussion
THP is the only oral medication to be proven effective in a double-blind placebo-controlled trial for dystonia (Fahn, 1983; Burke et al., 1986). Unfortunately, most patients cannot use THP due to intolerable side-effects at the high doses necessary for efficacy in dystonia (Schwarz and Bressman, 2009; Thenganatt and Jankovic, 2014). These side effects include cognitive impairment, constipation, dry mouth, urinary retention, and others (Burke et al., 1986; Jabbari et al., 1989; Guthrie et al., 2000; Lumsden et al., 2016). Here we used THP as a platform to further understand the mechanism of action of mAChR antagonists and identify specific mAChR subtypes that may serve as targets for more specific therapeutics with fewer side effects. We demonstrated that M4 mAChR located on striatal ChIs mediate, in part, the THP-induced increase in striatal DA release. Furthermore, we found that M1 mAChRs likely do not play a role in the effects of THP.
Our studies demonstrate that M1 mAChRs on neither dSPNs nor iSPNs contribute to the effects of THP in Tor1a+/+ or Tor1a+/ΔE KI mice. This is in contrast to a previous study that found elevated extracellular DA in the striatum in global M1 mAChR KO mice, albeit in an in vivo setting using microdialysis (Gerber et al., 2001b). This may reflect the fact that long loop connections are severed in our ex vivo slice preparation. Further, any local-circuit effects of M1 mAChR activation are likely to be indirect perhaps via SPN axon collateral synapses onto ChIs (Tepper et al., 2008). These connections might not be functional in our slice preparation or may be occluded by the local electrical stimulation used in our study.
Our results demonstrate that M4 mAChRs, in part, mediate the THP-induced rescue of DA release in Tor1a+/ΔE KI mice. KO of M4 mAChR from ChIs, but not dSPNs, diminished the increase in DA release caused by THP in both Tor1a+/+ and Tor1a+/ΔE KI mice. The residual effect of THP on DA release may be a result of THP acting on M2 mAChRs also located on striatal cholinergic interneurons, which are presumed to have an identical function to M4 mAChRs located on these cells (Threlfell et al., 2010). This is supported by the fact that KO of M4 from ChIs abolishes the effects of the M4 mAChR selective antagonist VU6021625. Surprisingly, KO of M4 mAChR from ChIs by itself did not normalize DA release as illustrated by the comparable baselines between control and KO mice. This may be due to homeostatic changes during development. In this context, the results from the antagonist experiments in intact adult mice are particularly important, because gene deletion experiments can be confounded by compensatory developmental changes in response to the knockout. Further, the fact that KO of M4 mAChR from ChI abolished the effects of VU6021625 in both Tor1a+/+ and Tor1a+/ΔE KI mice demonstrates the specificity of this compound. However, the response to the M4 mAChR antagonist in adult mice mirrored the response in the KO mice. Thus, the consistency of our results across both the M4 mAChR antagonist challenge in genetically unaltered adult mice and the conditional knockout experiments suggests that developmental confounds do not likely account for the results.
Although Tor1a+/ΔE KI mice do not exhibit a dystonic phenotype (Goodchild et al., 2005) these mice do exhibit a reduction in DA neurotransmission, a phenotype observed across many different forms of dystonia. Thus, the ability of THP and the subtype-selective mAChR antagonists to normalize striatal DA release in Tor1a+/ΔE KI mice represents a plausible mechanism of action for these compounds. Indeed, we have previously demonstrated that THP and the M4 mAChR subtype-selective antagonist VU6021625 ameliorates the dystonic movements exhibited by a knockin mouse model of DOPA-responsive dystonia, another model in which DA neurotransmission is significantly reduced (Rose et al., 2015; Moehle et al., 2020). Abnormalities in striatal DA neurotransmission have been implicated in diverse forms of dystonia including, adult-onset idiopathic dystonias, secondary dystonia due to Parkinson's disease or typical antipsychotic treatment, and inherited dystonia (Ichinose and Nagatsu, 1999; Tolosa and Compta, 2006; Carbon et al., 2009; Berman et al., 2013; Rilstone et al., 2013; Ng et al., 2014; Mehta et al., 2015). Thus, abnormal striatal DA neurotransmission is a convergent pathology across dystonias with diverse etiologies. While DA receptor agonists are generally not considered to be effective in dystonia, a recent reappraisal of these therapeutics suggests that both direct and indirect DA agonists are effective in subsets of dystonia patients and mouse models of dystonia (Fan et al., 2018). Further, unlike direct DA receptor agonists, which act at DA receptors in a temporally indiscriminate manner, THP and other mAChR antagonists provide the advantage of enhancing DA release while maintaining normal patterns of DA release encoded by DA neuron firing. However, mAChR antagonists may act through mechanisms in addition to DA neurotransmission to diminish dystonic symptoms. Previous studies have demonstrated that THP normalizes abnormal corticostriatal plasticity, including diminished long-term depression and loss of synaptic depotentiation in SPNs, observed in Tor1a+/ΔE KI mice via M1 mAChRs (Maltese et al., 2014; Martella et al., 2014). Thus, it is possible that a multiprong approach may be most effective for the amelioration of dystonia.
The role of ChIs in the mechanism of action of THP is consistent with the considerable evidence implicating ChI dysfunction in TOR1A dystonia. Normally, D2 DA receptor activation inhibits ChI firing rates, causing a reduction in extracellular ACh. In contrast, in Tor1a(ΔE) rodent models, the D2 DA receptor agonist quinpirole induces an increase in the ChI firing rate (Pisani et al., 2006; Martella et al., 2009; Grundmann et al., 2012; Sciamanna et al., 2012). Both THP and a more selective M2/M4 mAChR antagonist restore D2 DA receptor-mediated ChI responses in ex vivo brain slices of Tor1a+/ΔE KI mice (Scarduzio et al., 2017). Thus, THP and other more selective mAChR antagonists normalize both DA release and striatal ChI responses in Tor1a+/ΔE KI mice suggesting a central role for ChI in the development of novel therapeutics. However, it is not yet known if a selective M4 mAChR antagonist alone would similarly normalize ChI responses.
Taken together, our data demonstrate that THP normalizes DA release in Tor1a+/ΔE KI mice by blocking M4 mAChR on ChIs. This suggests that M4 subtype-selective mAChR antagonists, alone or in combination with other compounds, may be promising therapeutics for TOR1A dystonia. Further, because many of the dose-limiting peripheral side effects associated with THP, including constipation, dry mouth, and urinary retention, are mediated by M2 and M3 mAChR (Bymaster et al., 2003), M4 subtype-selective mAChR antagonists may improve dystonia while avoiding many of the side effects of the non-selective mAChR antagonists that are currently available to patients.
THP is the only oral medication to be proven effective in a double-blind placebo-controlled trial for dystonia (Fahn, 1983; Burke et al., 1986). Unfortunately, most patients cannot use THP due to intolerable side-effects at the high doses necessary for efficacy in dystonia (Schwarz and Bressman, 2009; Thenganatt and Jankovic, 2014). These side effects include cognitive impairment, constipation, dry mouth, urinary retention, and others (Burke et al., 1986; Jabbari et al., 1989; Guthrie et al., 2000; Lumsden et al., 2016). Here we used THP as a platform to further understand the mechanism of action of mAChR antagonists and identify specific mAChR subtypes that may serve as targets for more specific therapeutics with fewer side effects. We demonstrated that M4 mAChR located on striatal ChIs mediate, in part, the THP-induced increase in striatal DA release. Furthermore, we found that M1 mAChRs likely do not play a role in the effects of THP.
Our studies demonstrate that M1 mAChRs on neither dSPNs nor iSPNs contribute to the effects of THP in Tor1a+/+ or Tor1a+/ΔE KI mice. This is in contrast to a previous study that found elevated extracellular DA in the striatum in global M1 mAChR KO mice, albeit in an in vivo setting using microdialysis (Gerber et al., 2001b). This may reflect the fact that long loop connections are severed in our ex vivo slice preparation. Further, any local-circuit effects of M1 mAChR activation are likely to be indirect perhaps via SPN axon collateral synapses onto ChIs (Tepper et al., 2008). These connections might not be functional in our slice preparation or may be occluded by the local electrical stimulation used in our study.
Our results demonstrate that M4 mAChRs, in part, mediate the THP-induced rescue of DA release in Tor1a+/ΔE KI mice. KO of M4 mAChR from ChIs, but not dSPNs, diminished the increase in DA release caused by THP in both Tor1a+/+ and Tor1a+/ΔE KI mice. The residual effect of THP on DA release may be a result of THP acting on M2 mAChRs also located on striatal cholinergic interneurons, which are presumed to have an identical function to M4 mAChRs located on these cells (Threlfell et al., 2010). This is supported by the fact that KO of M4 from ChIs abolishes the effects of the M4 mAChR selective antagonist VU6021625. Surprisingly, KO of M4 mAChR from ChIs by itself did not normalize DA release as illustrated by the comparable baselines between control and KO mice. This may be due to homeostatic changes during development. In this context, the results from the antagonist experiments in intact adult mice are particularly important, because gene deletion experiments can be confounded by compensatory developmental changes in response to the knockout. Further, the fact that KO of M4 mAChR from ChI abolished the effects of VU6021625 in both Tor1a+/+ and Tor1a+/ΔE KI mice demonstrates the specificity of this compound. However, the response to the M4 mAChR antagonist in adult mice mirrored the response in the KO mice. Thus, the consistency of our results across both the M4 mAChR antagonist challenge in genetically unaltered adult mice and the conditional knockout experiments suggests that developmental confounds do not likely account for the results.
Although Tor1a+/ΔE KI mice do not exhibit a dystonic phenotype (Goodchild et al., 2005) these mice do exhibit a reduction in DA neurotransmission, a phenotype observed across many different forms of dystonia. Thus, the ability of THP and the subtype-selective mAChR antagonists to normalize striatal DA release in Tor1a+/ΔE KI mice represents a plausible mechanism of action for these compounds. Indeed, we have previously demonstrated that THP and the M4 mAChR subtype-selective antagonist VU6021625 ameliorates the dystonic movements exhibited by a knockin mouse model of DOPA-responsive dystonia, another model in which DA neurotransmission is significantly reduced (Rose et al., 2015; Moehle et al., 2020). Abnormalities in striatal DA neurotransmission have been implicated in diverse forms of dystonia including, adult-onset idiopathic dystonias, secondary dystonia due to Parkinson's disease or typical antipsychotic treatment, and inherited dystonia (Ichinose and Nagatsu, 1999; Tolosa and Compta, 2006; Carbon et al., 2009; Berman et al., 2013; Rilstone et al., 2013; Ng et al., 2014; Mehta et al., 2015). Thus, abnormal striatal DA neurotransmission is a convergent pathology across dystonias with diverse etiologies. While DA receptor agonists are generally not considered to be effective in dystonia, a recent reappraisal of these therapeutics suggests that both direct and indirect DA agonists are effective in subsets of dystonia patients and mouse models of dystonia (Fan et al., 2018). Further, unlike direct DA receptor agonists, which act at DA receptors in a temporally indiscriminate manner, THP and other mAChR antagonists provide the advantage of enhancing DA release while maintaining normal patterns of DA release encoded by DA neuron firing. However, mAChR antagonists may act through mechanisms in addition to DA neurotransmission to diminish dystonic symptoms. Previous studies have demonstrated that THP normalizes abnormal corticostriatal plasticity, including diminished long-term depression and loss of synaptic depotentiation in SPNs, observed in Tor1a+/ΔE KI mice via M1 mAChRs (Maltese et al., 2014; Martella et al., 2014). Thus, it is possible that a multiprong approach may be most effective for the amelioration of dystonia.
The role of ChIs in the mechanism of action of THP is consistent with the considerable evidence implicating ChI dysfunction in TOR1A dystonia. Normally, D2 DA receptor activation inhibits ChI firing rates, causing a reduction in extracellular ACh. In contrast, in Tor1a(ΔE) rodent models, the D2 DA receptor agonist quinpirole induces an increase in the ChI firing rate (Pisani et al., 2006; Martella et al., 2009; Grundmann et al., 2012; Sciamanna et al., 2012). Both THP and a more selective M2/M4 mAChR antagonist restore D2 DA receptor-mediated ChI responses in ex vivo brain slices of Tor1a+/ΔE KI mice (Scarduzio et al., 2017). Thus, THP and other more selective mAChR antagonists normalize both DA release and striatal ChI responses in Tor1a+/ΔE KI mice suggesting a central role for ChI in the development of novel therapeutics. However, it is not yet known if a selective M4 mAChR antagonist alone would similarly normalize ChI responses.
Taken together, our data demonstrate that THP normalizes DA release in Tor1a+/ΔE KI mice by blocking M4 mAChR on ChIs. This suggests that M4 subtype-selective mAChR antagonists, alone or in combination with other compounds, may be promising therapeutics for TOR1A dystonia. Further, because many of the dose-limiting peripheral side effects associated with THP, including constipation, dry mouth, and urinary retention, are mediated by M2 and M3 mAChR (Bymaster et al., 2003), M4 subtype-selective mAChR antagonists may improve dystonia while avoiding many of the side effects of the non-selective mAChR antagonists that are currently available to patients.
CRediT authorship contribution statement
Anthony M. Downs: Conceptualization, Formal analysis, Investigation, Writing – original draft, Visualization, Funding acquisition, Writing – review & editing. Yuping Donsante: Investigation, Writing – review & editing. H.A. Jinnah: Conceptualization, Writing – review & editing. Ellen J. Hess: Conceptualization, Formal analysis, Writing – original draft, Visualization, Funding acquisition, Writing – review & editing.
Acknowledgments
Funding: This work was supported by United States Department of Defense grants [W81XWH-15-1-0545] and [W81XWH-20-1-0446]; United States National Institute of Health Grants [F31 NS103363] and [T32 GM008602]; and Cure Dystonia Now.
This research project was supported in part by the Emory University Integrated Cellular Imaging Core. This study was also supported in part by the Emory HPLC Bioanalytical Core (EHBC), which was supported by the Department of Pharmacology and Chemical Biology, Emory University School of Medicine, and the Georgia Clinical & Translational Science Alliance of the National Institutes of Health under Award Number UL1TR002378.
References
Alcantara et al., 2001
A.A. Alcantara, L. Mrzljak, R.L. Jakab, A.I. Levey, S.M. Hersch, P.S. Goldman-Rakic
Muscarinic m1 and m2 receptor proteins in local circuit and projection neurons of the primate striatum: anatomical evidence for cholinergic modulation of glutamatergic prefronto-striatal pathways
J. Comp. Neurol., 434 (2001), pp. 445-460
Asanuma et al., 2005
K. Asanuma, Y. Ma, J. Okulski, V. Dhawan, T. Chaly, M. Carbon, S.B. Bressman, D. Eidelberg
Decreased striatal D2 receptor binding in non-manifesting carriers of the DYT1 dystonia mutation
Neurology, 64 (2005), pp. 347-349
Augood et al., 2004
S.J. Augood, Z. Hollingsworth, D.S. Albers, L. Yang, J. Leung, X.O. Breakefield, D.G. Standaert
Dopamine transmission in DYT1 dystonia
Adv. Neurol., 94 (2004), pp. 53-60
Balcioglu et al., 2007
A. Balcioglu, M.O. Kim, N. Sharma, J.H. Cha, X.O. Breakefield, D.G. Standaert
Dopamine release is impaired in a mouse model of DYT1 dystonia
J. Neurochem., 102 (2007), pp. 783-788
View PDF
Berman et al., 2013
B.D. Berman, M. Hallett, P. Herscovitch, K. Simonyan
Striatal dopaminergic dysfunction at rest and during task performance in writer’s cramp
Brain, 136 (2013), pp. 3645-3658
View PDF
Bolden et al., 1992
C. Bolden, B. Cusack, E. Richelson
Antagonism by antimuscarinic and neuroleptic compounds at the five cloned human muscarinic cholinergic receptors expressed in Chinese hamster ovary cells
J. Pharmacol. Exp. Ther., 260 (1992), pp. 576-580
Buckley et al., 1988
N.J. Buckley, T.I. Bonner, M.R. Brann
Localization of a family of muscarinic receptor mRNAs in rat brain
J. Neurosci., 8 (1988), pp. 4646-4652
View PDF
Burke et al., 1986
R.E. Burke, S. Fahn, C.D. Marsden
Torsion dystonia: a double-blind, prospective trial of high-dosage trihexyphenidyl
Neurology, 36 (1986), pp. 160-164
Bymaster et al., 2003
F.P. Bymaster, P.A. Carter, M. Yamada, J. Gomeza, J. Wess, S.E. Hamilton, N.M. Nathanson, D.L. McKinzie, C.C. Felder
Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity
Eur. J. Neurosci., 17 (2003), pp. 1403-1410
Carbon et al., 2009
M. Carbon, M. Niethammer, S. Peng, D. Raymond, V. Dhawan, T. Chaly, Y. Ma, S. Bressman, D. Eidelberg
Abnormal striatal and thalamic dopamine neurotransmission: genotype-related features of dystonia
Neurology, 72 (2009), pp. 2097-2103
Dorje et al., 1991
F. Dorje, J. Wess, G. Lambrecht, R. Tacke, E. Mutschler, M.R. Brann
Antagonist binding profiles of five cloned human muscarinic receptor subtypes
J. Pharmacol. Exp. Ther., 256 (1991), pp. 727-733
Downs et al., 2019
A.M. Downs, X. Fan, C. Donsante, H.A. Jinnah, E.J. Hess
Trihexyphenidyl rescues the deficit in dopamine neurotransmission in a mouse model of DYT1 dystonia
Neurobiol. Dis., 125 (2019), pp. 115-122
Exley and Cragg, 2008
R. Exley, S.J. Cragg
Presynaptic nicotinic receptors: a dynamic and diverse cholinergic filter of striatal dopamine neurotransmission
Br. J. Pharmacol., 153 (Suppl. 1) (2008), pp. S283-S297
View PDF
Fahn, 1983
S. Fahn
High-dosage anticholinergic therapy in dystonia
Adv. Neurol., 37 (1983), pp. 177-188
Fan et al., 2018
X. Fan, Y. Donsante, H.A. Jinnah, E.J. Hess
Dopamine receptor agonist treatment of idiopathic dystonia: a reappraisal in humans and mice
J. Pharmacol. Exp. Ther., 365 (2018), pp. 20-26
View PDF
Foster et al., 2014
D.J. Foster, P.R. Gentry, J.E. Lizardi-Ortiz, T.M. Bridges, M.R. Wood, C.M. Niswender, D. Sulzer, C.W. Lindsley, Z. Xiang, P.J. Conn
M5 receptor activation produces opposing physiological outcomes in dopamine neurons depending on the receptor’s location
J. Neurosci., 34 (2014), pp. 3253-3262
Foster et al., 2016
D.J. Foster, J.M. Wilson, D.H. Remke, M.S. Mahmood, M.J. Uddin, J. Wess, S. Patel, L.J. Marnett, C.M. Niswender, C.K. Jones, Z. Xiang, C.W. Lindsley, J.M. Rook, P.J. Conn
Antipsychotic-like effects of M4 positive allosteric modulators are mediated by CB2 receptor-dependent inhibition of dopamine release
Neuron, 91 (2016), pp. 1244-1252
Gerber et al., 2001a
D.J. Gerber, T.D. Sotnikova, R.R. Gainetdinov, S.Y. Huang, M.G. Caron, S. Tonegawa
Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice
Proc. Natl. Acad. Sci., 98 (2001), pp. 15312-15317
Gerber et al., 2001b
D.J. Gerber, T.D. Sotnikova, R.R. Gainetdinov, S.Y. Huang, M.G. Caron, S. Tonegawa
Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice
Proc. Natl. Acad. Sci. U. S. A., 98 (2001), pp. 15312-15317
Goodchild et al., 2005
R.E. Goodchild, C.E. Kim, W.T. Dauer
Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope
Neuron, 48 (2005), pp. 923-932
Grundmann et al., 2012
K. Grundmann, N. Glöckle, G. Martella, G. Sciamanna, T.K. Hauser, L. Yu, S. Castaneda, B. Pichler, B. Fehrenbacher, M. Schaller, B. Nuscher, C. Haass, J. Hettich, Z. Yue, H.P. Nguyen, A. Pisani, O. Riess, T. Ott
Generation of a novel rodent model for DYT1 dystonia
Neurobiol. Dis., 47 (2012), pp. 61-74
Guthrie et al., 2000
S.K. Guthrie, L. Manzey, D. Scott, B. Giordani, R. Tandon
Comparison of central and peripheral pharmacologic effects of biperiden and trihexyphenidyl in human volunteers
J. Clin. Psychopharmacol., 20 (2000), pp. 77-83
Harrison et al., 1996
M.B. Harrison, M. Tissot, R.G. Wiley
Expression of m1 and m4 muscarinic receptor mRNA in the striatum following a selective lesion of striatonigral neurons
Brain Res., 734 (1996), pp. 323-326
Hernandez-Flores et al., 2015
T. Hernandez-Flores, O. Hernandez-Gonzalez, M.B. Perez-Ramirez, E. Lara-Gonzalez, M.A. Arias-Garcia, M. Duhne, A. Perez-Burgos, G.A. Prieto, A. Figueroa, E. Galarraga, J. Bargas
Modulation of direct pathway striatal projection neurons by muscarinic M(4)-type receptors
Neuropharmacology, 89 (2015), pp. 232-244
Hintiryan et al., 2016
H. Hintiryan, N.N. Foster, I. Bowman, M. Bay, M.Y. Song, L. Gou, S. Yamashita, M.S. Bienkowski, B. Zingg, M. Zhu, X.W. Yang, J.C. Shih, A.W. Toga, H.W. Dong
The mouse cortico-striatal projectome
Nat. Neurosci., 19 (2016), pp. 1100-1114
Ichinose and Nagatsu, 1999
H. Ichinose, T. Nagatsu
Molecular genetics of DOPA-responsive dystonia
Adv. Neurol., 80 (1999), pp. 195-198
Jabbari et al., 1989
B. Jabbari, B. Scherokman, C.H. Gunderson, M.L. Rosenberg, J. Miller
Treatment of movement disorders with trihexyphenidyl
Mov. Disord., 4 (1989), pp. 202-212
Jeon et al., 2010
J. Jeon, D. Dencker, G. Wörtwein, D.P. Woldbye, Y. Cui, A.A. Davis, A.I. Levey, G. Schütz, T.N. Sager, A. Mørk, C. Li, C.X. Deng, A. Fink-Jensen, J. Wess
A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors
J. Neurosci., 30 (2010), pp. 2396-2405
Kamsler et al., 2010
A. Kamsler, T.J. McHugh, D. Gerber, S.Y. Huang, S. Tonegawa
Presynaptic m1 muscarinic receptors are necessary for mGluR long-term depression in the hippocampus
Proc. Natl. Acad. Sci. U. S. A., 107 (2010), pp. 1618-1623
Levey et al., 1991
A.I. Levey, C.A. Kitt, W.F. Simonds, D.L. Price, M.R. Brann
Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies
J. Neurosci., 11 (1991), pp. 3218-3226
Lumsden et al., 2016
D.E. Lumsden, M. Kaminska, S. Tomlin, J.P. Lin
Medication use in childhood dystonia
Eur. J. Paediatr. Neurol., 20 (2016), pp. 625-629
Maltese et al., 2014
M. Maltese, G. Martella, G. Madeo, I. Fagiolo, A. Tassone, G. Ponterio, G. Sciamanna, P. Burbaud, P.J. Conn, P. Bonsi, A. Pisani
Anticholinergic drugs rescue synaptic plasticity in DYT1 dystonia: role of M1 muscarinic receptors
Mov. Disord., 29 (2014), pp. 1655-1665
Martella et al., 2009
G. Martella, A. Tassone, G. Sciamanna, P. Platania, D. Cuomo, M.T. Viscomi, P. Bonsi, E. Cacci, S. Biagioni, A. Usiello, G. Bernardi, N. Sharma, D.G. Standaert, A. Pisani
Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine
Brain, 132 (2009), pp. 2336-2349
Martella et al., 2014
G. Martella, M. Maltese, R. Nistico, T. Schirinzi, G. Madeo, G. Sciamanna, G. Ponterio, A. Tassone, G. Mandolesi, V. Vanni, M. Pignatelli, P. Bonsi, A. Pisani
Regional specificity of synaptic plasticity deficits in a knock-in mouse model of DYT1 dystonia
Neurobiol. Dis., 65 (2014), pp. 124-132
Mehta et al., 2015
S.H. Mehta, J.C. Morgan, K.D. Sethi
Drug-induced movement disorders
Neurol. Clin., 33 (2015), pp. 153-174
Moehle et al., 2020
M.S. Moehle, A.M. Bender, J.W. Dickerson, D.J. Foster, Y. Donsante, W. Peng, Z. Bryant, T.M. Bridges, S. Chang, K.J. Watson, J.C. O’Neill, J.L. Engers, L. Peng, A.L. Rodriguez, C.M. Niswender, C.W. Lindsley, E.J. Hess, P.J. Conn, J.M. Rook
Discovery of the First Selective M4 Muscarinic Acetylcholine Receptor Antagonists with “In Vivo” Anti-Parkinsonian and Anti-Dystonic Efficacy
(2020)
bioRxiv:2020.2010.2012.324152
Nair et al., 2015
A.G. Nair, O. Gutierrez-Arenas, O. Eriksson, P. Vincent, J. Hellgren Kotaleski
Sensing positive versus negative reward signals through adenylyl cyclase-coupled GPCRs in direct and indirect pathway striatal medium spiny neurons
J. Neurosci., 35 (2015), pp. 14017-14030
Nair et al., 2019
A.G. Nair, L.R.V. Castro, M. El Khoury, V. Gorgievski, B. Giros, E.T. Tzavara, J. Hellgren-Kotaleski, P. Vincent
The high efficacy of muscarinic M4 receptor in D1 medium spiny neurons reverses striatal hyperdopaminergia
Neuropharmacology, 146 (2019), pp. 74-83
Ng et al., 2014
J. Ng, et al.
Dopamine transporter deficiency syndrome: phenotypic spectrum from infancy to adulthood
Brain, 137 (2014), pp. 1107-1119
Ozelius et al., 1997
L.J. Ozelius, J.W. Hewett, C.E. Page, S.B. Bressman, P.L. Kramer, C. Shalish, D. de Leon, M.F. Brin, D. Raymond, D.P. Corey, S. Fahn, N.J. Risch, A.J. Buckler, J.F. Gusella, X.O. Breakefield
The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein
Nat. Genet., 17 (1997), pp. 40-48
Page et al., 2010
M.E. Page, L. Bao, P. Andre, J. Pelta-Heller, E. Sluzas, P. Gonzalez-Alegre, A. Bogush, L.E. Khan, L. Iacovitti, M.E. Rice, M.E. Ehrlich
Cell-autonomous alteration of dopaminergic transmission by wild type and mutant (DeltaE) TorsinA in transgenic mice
Neurobiol. Dis., 39 (2010), pp. 318-326
Pancani et al., 2014
T. Pancani, C. Bolarinwa, Y. Smith, C.W. Lindsley, P.J. Conn, Z. Xiang
M4 mAChR-mediated modulation of glutamatergic transmission at corticostriatal synapses
ACS Chem. Neurosci., 5 (2014), pp. 318-324
Pisani et al., 2006
A. Pisani, G. Martella, A. Tscherter, P. Bonsi, N. Sharma, G. Bernardi, D.G. Standaert
Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia
Neurobiol. Dis., 24 (2006), pp. 318-325
Rilstone et al., 2013
J.J. Rilstone, R.A. Alkhater, B.A. Minassian
Brain dopamine-serotonin vesicular transport disease and its treatment
N. Engl. J. Med., 368 (2013), pp. 543-550
Rose et al., 2015
S.J. Rose, X.Y. Yu, A.K. Heinzer, P. Harrast, X. Fan, R.S. Raike, V.B. Thompson, J.F. Pare, D. Weinshenker, Y. Smith, H.A. Jinnah, E.J. Hess
A new knock-in mouse model of l-DOPA-responsive dystonia
Brain, 138 (2015), pp. 2987-3002
Scarduzio et al., 2017
M. Scarduzio, C.N. Zimmerman, K.L. Jaunarajs, Q. Wang, D.G. Standaert, L.L. McMahon
Strength of cholinergic tone dictates the polarity of dopamine D2 receptor modulation of striatal cholinergic interneuron excitability in DYT1 dystonia
Exp. Neurol., 295 (2017), pp. 162-175
Schwarz and Bressman, 2009
C.S. Schwarz, S.B. Bressman
Genetics and treatment of dystonia
Neurol. Clin., 27 (2009), pp. 697-718
vi
Sciamanna et al., 2012
G. Sciamanna, A. Tassone, G. Mandolesi, F. Puglisi, G. Ponterio, G. Martella, G. Madeo, G. Bernardi, D.G. Standaert, P. Bonsi, A. Pisani
Cholinergic dysfunction alters synaptic integration between thalamostriatal and corticostriatal inputs in DYT1 dystonia
J. Neurosci., 32 (2012), pp. 11991-12004
Song et al., 2012
C.H. Song, X. Fan, C.J. Exeter, E.J. Hess, H.A. Jinnah
Functional analysis of dopaminergic systems in a DYT1 knock-in mouse model of dystonia
Neurobiol. Dis., 48 (2012), pp. 66-78
Tepper et al., 2008
J.M. Tepper, C.J. Wilson, T. Koos
Feedforward and feedback inhibition in neostriatal GABAergic spiny neurons
Brain Res. Rev., 58 (2008), pp. 272-281
Thenganatt and Jankovic, 2014
M.A. Thenganatt, J. Jankovic
Treatment of dystonia
Neurotherapeutics, 11 (2014), pp. 139-152
Threlfell and Cragg, 2011
S. Threlfell, S.J. Cragg
Dopamine signaling in dorsal versus ventral striatum: the dynamic role of cholinergic interneurons
Front. Syst. Neurosci., 5 (2011), p. 11
Threlfell et al., 2010
S. Threlfell, M.A. Clements, T. Khodai, I.S. Pienaar, R. Exley, J. Wess, S.J. Cragg
Striatal muscarinic receptors promote activity dependence of dopamine transmission via distinct receptor subtypes on cholinergic interneurons in ventral versus dorsal striatum
J. Neurosci., 30 (2010), pp. 3398-3408
Tolosa and Compta, 2006
E. Tolosa, Y. Compta
Dystonia in Parkinson’s disease
J. Neurol., 253 (Suppl. 7) (2006)
Vii7-13
Yamada et al., 2003
M. Yamada, A.S. Basile, I. Fedorova, W. Zhang, A. Duttaroy, Y. Cui, K.G. Lamping, F.M. Faraci, C.X. Deng, J. Wess
Novel insights into M5 muscarinic acetylcholine receptor function by the use of gene targeting technology
Life Sci., 74 (2003), pp. 345-353
Yorgason et al., 2011
J.T. Yorgason, R.A. Espana, S.R. Jones
Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures
J. Neurosci. Methods, 202 (2011), pp. 158-164
Zhang et al., 2002a
W. Zhang, M. Yamada, J. Gomeza, A.S. Basile, J. Wess
Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1-M5 muscarinic receptor knock-out mice
J. Neurosci., 22 (2002), pp. 6347-6352
Zhang et al., 2002b
W. Zhang, A.S. Basile, J. Gomeza, L.A. Volpicelli, A.I. Levey, J. Wess
Characterization of central inhibitory muscarinic autoreceptors by the use of muscarinic acetylcholine receptor knock-out mice
J. Neurosci., 22 (2002), pp. 1709-1717
Zhang et al., 2009a
L. Zhang, W.M. Doyon, J.J. Clark, P.E. Phillips, J.A. Dani
Controls of tonic and phasic dopamine transmission in the dorsal and ventral striatum
Mol. Pharmacol., 76 (2009), pp. 396-404
Zhang et al., 2009b
T. Zhang, L. Zhang, Y. Liang, A.G. Siapas, F.M. Zhou, J.A. Dani
Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine
J. Neurosci., 29 (2009), pp. 4035-4043
Anthony M. Downs: Conceptualization, Formal analysis, Investigation, Writing – original draft, Visualization, Funding acquisition, Writing – review & editing. Yuping Donsante: Investigation, Writing – review & editing. H.A. Jinnah: Conceptualization, Writing – review & editing. Ellen J. Hess: Conceptualization, Formal analysis, Writing – original draft, Visualization, Funding acquisition, Writing – review & editing.
Acknowledgments
Funding: This work was supported by United States Department of Defense grants [W81XWH-15-1-0545] and [W81XWH-20-1-0446]; United States National Institute of Health Grants [F31 NS103363] and [T32 GM008602]; and Cure Dystonia Now.
This research project was supported in part by the Emory University Integrated Cellular Imaging Core. This study was also supported in part by the Emory HPLC Bioanalytical Core (EHBC), which was supported by the Department of Pharmacology and Chemical Biology, Emory University School of Medicine, and the Georgia Clinical & Translational Science Alliance of the National Institutes of Health under Award Number UL1TR002378.
References
Alcantara et al., 2001
A.A. Alcantara, L. Mrzljak, R.L. Jakab, A.I. Levey, S.M. Hersch, P.S. Goldman-Rakic
Muscarinic m1 and m2 receptor proteins in local circuit and projection neurons of the primate striatum: anatomical evidence for cholinergic modulation of glutamatergic prefronto-striatal pathways
J. Comp. Neurol., 434 (2001), pp. 445-460
Asanuma et al., 2005
K. Asanuma, Y. Ma, J. Okulski, V. Dhawan, T. Chaly, M. Carbon, S.B. Bressman, D. Eidelberg
Decreased striatal D2 receptor binding in non-manifesting carriers of the DYT1 dystonia mutation
Neurology, 64 (2005), pp. 347-349
Augood et al., 2004
S.J. Augood, Z. Hollingsworth, D.S. Albers, L. Yang, J. Leung, X.O. Breakefield, D.G. Standaert
Dopamine transmission in DYT1 dystonia
Adv. Neurol., 94 (2004), pp. 53-60
Balcioglu et al., 2007
A. Balcioglu, M.O. Kim, N. Sharma, J.H. Cha, X.O. Breakefield, D.G. Standaert
Dopamine release is impaired in a mouse model of DYT1 dystonia
J. Neurochem., 102 (2007), pp. 783-788
View PDF
Berman et al., 2013
B.D. Berman, M. Hallett, P. Herscovitch, K. Simonyan
Striatal dopaminergic dysfunction at rest and during task performance in writer’s cramp
Brain, 136 (2013), pp. 3645-3658
View PDF
Bolden et al., 1992
C. Bolden, B. Cusack, E. Richelson
Antagonism by antimuscarinic and neuroleptic compounds at the five cloned human muscarinic cholinergic receptors expressed in Chinese hamster ovary cells
J. Pharmacol. Exp. Ther., 260 (1992), pp. 576-580
Buckley et al., 1988
N.J. Buckley, T.I. Bonner, M.R. Brann
Localization of a family of muscarinic receptor mRNAs in rat brain
J. Neurosci., 8 (1988), pp. 4646-4652
View PDF
Burke et al., 1986
R.E. Burke, S. Fahn, C.D. Marsden
Torsion dystonia: a double-blind, prospective trial of high-dosage trihexyphenidyl
Neurology, 36 (1986), pp. 160-164
Bymaster et al., 2003
F.P. Bymaster, P.A. Carter, M. Yamada, J. Gomeza, J. Wess, S.E. Hamilton, N.M. Nathanson, D.L. McKinzie, C.C. Felder
Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity
Eur. J. Neurosci., 17 (2003), pp. 1403-1410
Carbon et al., 2009
M. Carbon, M. Niethammer, S. Peng, D. Raymond, V. Dhawan, T. Chaly, Y. Ma, S. Bressman, D. Eidelberg
Abnormal striatal and thalamic dopamine neurotransmission: genotype-related features of dystonia
Neurology, 72 (2009), pp. 2097-2103
Dorje et al., 1991
F. Dorje, J. Wess, G. Lambrecht, R. Tacke, E. Mutschler, M.R. Brann
Antagonist binding profiles of five cloned human muscarinic receptor subtypes
J. Pharmacol. Exp. Ther., 256 (1991), pp. 727-733
Downs et al., 2019
A.M. Downs, X. Fan, C. Donsante, H.A. Jinnah, E.J. Hess
Trihexyphenidyl rescues the deficit in dopamine neurotransmission in a mouse model of DYT1 dystonia
Neurobiol. Dis., 125 (2019), pp. 115-122
Exley and Cragg, 2008
R. Exley, S.J. Cragg
Presynaptic nicotinic receptors: a dynamic and diverse cholinergic filter of striatal dopamine neurotransmission
Br. J. Pharmacol., 153 (Suppl. 1) (2008), pp. S283-S297
View PDF
Fahn, 1983
S. Fahn
High-dosage anticholinergic therapy in dystonia
Adv. Neurol., 37 (1983), pp. 177-188
Fan et al., 2018
X. Fan, Y. Donsante, H.A. Jinnah, E.J. Hess
Dopamine receptor agonist treatment of idiopathic dystonia: a reappraisal in humans and mice
J. Pharmacol. Exp. Ther., 365 (2018), pp. 20-26
View PDF
Foster et al., 2014
D.J. Foster, P.R. Gentry, J.E. Lizardi-Ortiz, T.M. Bridges, M.R. Wood, C.M. Niswender, D. Sulzer, C.W. Lindsley, Z. Xiang, P.J. Conn
M5 receptor activation produces opposing physiological outcomes in dopamine neurons depending on the receptor’s location
J. Neurosci., 34 (2014), pp. 3253-3262
Foster et al., 2016
D.J. Foster, J.M. Wilson, D.H. Remke, M.S. Mahmood, M.J. Uddin, J. Wess, S. Patel, L.J. Marnett, C.M. Niswender, C.K. Jones, Z. Xiang, C.W. Lindsley, J.M. Rook, P.J. Conn
Antipsychotic-like effects of M4 positive allosteric modulators are mediated by CB2 receptor-dependent inhibition of dopamine release
Neuron, 91 (2016), pp. 1244-1252
Gerber et al., 2001a
D.J. Gerber, T.D. Sotnikova, R.R. Gainetdinov, S.Y. Huang, M.G. Caron, S. Tonegawa
Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice
Proc. Natl. Acad. Sci., 98 (2001), pp. 15312-15317
Gerber et al., 2001b
D.J. Gerber, T.D. Sotnikova, R.R. Gainetdinov, S.Y. Huang, M.G. Caron, S. Tonegawa
Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice
Proc. Natl. Acad. Sci. U. S. A., 98 (2001), pp. 15312-15317
Goodchild et al., 2005
R.E. Goodchild, C.E. Kim, W.T. Dauer
Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope
Neuron, 48 (2005), pp. 923-932
Grundmann et al., 2012
K. Grundmann, N. Glöckle, G. Martella, G. Sciamanna, T.K. Hauser, L. Yu, S. Castaneda, B. Pichler, B. Fehrenbacher, M. Schaller, B. Nuscher, C. Haass, J. Hettich, Z. Yue, H.P. Nguyen, A. Pisani, O. Riess, T. Ott
Generation of a novel rodent model for DYT1 dystonia
Neurobiol. Dis., 47 (2012), pp. 61-74
Guthrie et al., 2000
S.K. Guthrie, L. Manzey, D. Scott, B. Giordani, R. Tandon
Comparison of central and peripheral pharmacologic effects of biperiden and trihexyphenidyl in human volunteers
J. Clin. Psychopharmacol., 20 (2000), pp. 77-83
Harrison et al., 1996
M.B. Harrison, M. Tissot, R.G. Wiley
Expression of m1 and m4 muscarinic receptor mRNA in the striatum following a selective lesion of striatonigral neurons
Brain Res., 734 (1996), pp. 323-326
Hernandez-Flores et al., 2015
T. Hernandez-Flores, O. Hernandez-Gonzalez, M.B. Perez-Ramirez, E. Lara-Gonzalez, M.A. Arias-Garcia, M. Duhne, A. Perez-Burgos, G.A. Prieto, A. Figueroa, E. Galarraga, J. Bargas
Modulation of direct pathway striatal projection neurons by muscarinic M(4)-type receptors
Neuropharmacology, 89 (2015), pp. 232-244
Hintiryan et al., 2016
H. Hintiryan, N.N. Foster, I. Bowman, M. Bay, M.Y. Song, L. Gou, S. Yamashita, M.S. Bienkowski, B. Zingg, M. Zhu, X.W. Yang, J.C. Shih, A.W. Toga, H.W. Dong
The mouse cortico-striatal projectome
Nat. Neurosci., 19 (2016), pp. 1100-1114
Ichinose and Nagatsu, 1999
H. Ichinose, T. Nagatsu
Molecular genetics of DOPA-responsive dystonia
Adv. Neurol., 80 (1999), pp. 195-198
Jabbari et al., 1989
B. Jabbari, B. Scherokman, C.H. Gunderson, M.L. Rosenberg, J. Miller
Treatment of movement disorders with trihexyphenidyl
Mov. Disord., 4 (1989), pp. 202-212
Jeon et al., 2010
J. Jeon, D. Dencker, G. Wörtwein, D.P. Woldbye, Y. Cui, A.A. Davis, A.I. Levey, G. Schütz, T.N. Sager, A. Mørk, C. Li, C.X. Deng, A. Fink-Jensen, J. Wess
A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors
J. Neurosci., 30 (2010), pp. 2396-2405
Kamsler et al., 2010
A. Kamsler, T.J. McHugh, D. Gerber, S.Y. Huang, S. Tonegawa
Presynaptic m1 muscarinic receptors are necessary for mGluR long-term depression in the hippocampus
Proc. Natl. Acad. Sci. U. S. A., 107 (2010), pp. 1618-1623
Levey et al., 1991
A.I. Levey, C.A. Kitt, W.F. Simonds, D.L. Price, M.R. Brann
Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies
J. Neurosci., 11 (1991), pp. 3218-3226
Lumsden et al., 2016
D.E. Lumsden, M. Kaminska, S. Tomlin, J.P. Lin
Medication use in childhood dystonia
Eur. J. Paediatr. Neurol., 20 (2016), pp. 625-629
Maltese et al., 2014
M. Maltese, G. Martella, G. Madeo, I. Fagiolo, A. Tassone, G. Ponterio, G. Sciamanna, P. Burbaud, P.J. Conn, P. Bonsi, A. Pisani
Anticholinergic drugs rescue synaptic plasticity in DYT1 dystonia: role of M1 muscarinic receptors
Mov. Disord., 29 (2014), pp. 1655-1665
Martella et al., 2009
G. Martella, A. Tassone, G. Sciamanna, P. Platania, D. Cuomo, M.T. Viscomi, P. Bonsi, E. Cacci, S. Biagioni, A. Usiello, G. Bernardi, N. Sharma, D.G. Standaert, A. Pisani
Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine
Brain, 132 (2009), pp. 2336-2349
Martella et al., 2014
G. Martella, M. Maltese, R. Nistico, T. Schirinzi, G. Madeo, G. Sciamanna, G. Ponterio, A. Tassone, G. Mandolesi, V. Vanni, M. Pignatelli, P. Bonsi, A. Pisani
Regional specificity of synaptic plasticity deficits in a knock-in mouse model of DYT1 dystonia
Neurobiol. Dis., 65 (2014), pp. 124-132
Mehta et al., 2015
S.H. Mehta, J.C. Morgan, K.D. Sethi
Drug-induced movement disorders
Neurol. Clin., 33 (2015), pp. 153-174
Moehle et al., 2020
M.S. Moehle, A.M. Bender, J.W. Dickerson, D.J. Foster, Y. Donsante, W. Peng, Z. Bryant, T.M. Bridges, S. Chang, K.J. Watson, J.C. O’Neill, J.L. Engers, L. Peng, A.L. Rodriguez, C.M. Niswender, C.W. Lindsley, E.J. Hess, P.J. Conn, J.M. Rook
Discovery of the First Selective M4 Muscarinic Acetylcholine Receptor Antagonists with “In Vivo” Anti-Parkinsonian and Anti-Dystonic Efficacy
(2020)
bioRxiv:2020.2010.2012.324152
Nair et al., 2015
A.G. Nair, O. Gutierrez-Arenas, O. Eriksson, P. Vincent, J. Hellgren Kotaleski
Sensing positive versus negative reward signals through adenylyl cyclase-coupled GPCRs in direct and indirect pathway striatal medium spiny neurons
J. Neurosci., 35 (2015), pp. 14017-14030
Nair et al., 2019
A.G. Nair, L.R.V. Castro, M. El Khoury, V. Gorgievski, B. Giros, E.T. Tzavara, J. Hellgren-Kotaleski, P. Vincent
The high efficacy of muscarinic M4 receptor in D1 medium spiny neurons reverses striatal hyperdopaminergia
Neuropharmacology, 146 (2019), pp. 74-83
Ng et al., 2014
J. Ng, et al.
Dopamine transporter deficiency syndrome: phenotypic spectrum from infancy to adulthood
Brain, 137 (2014), pp. 1107-1119
Ozelius et al., 1997
L.J. Ozelius, J.W. Hewett, C.E. Page, S.B. Bressman, P.L. Kramer, C. Shalish, D. de Leon, M.F. Brin, D. Raymond, D.P. Corey, S. Fahn, N.J. Risch, A.J. Buckler, J.F. Gusella, X.O. Breakefield
The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein
Nat. Genet., 17 (1997), pp. 40-48
Page et al., 2010
M.E. Page, L. Bao, P. Andre, J. Pelta-Heller, E. Sluzas, P. Gonzalez-Alegre, A. Bogush, L.E. Khan, L. Iacovitti, M.E. Rice, M.E. Ehrlich
Cell-autonomous alteration of dopaminergic transmission by wild type and mutant (DeltaE) TorsinA in transgenic mice
Neurobiol. Dis., 39 (2010), pp. 318-326
Pancani et al., 2014
T. Pancani, C. Bolarinwa, Y. Smith, C.W. Lindsley, P.J. Conn, Z. Xiang
M4 mAChR-mediated modulation of glutamatergic transmission at corticostriatal synapses
ACS Chem. Neurosci., 5 (2014), pp. 318-324
Pisani et al., 2006
A. Pisani, G. Martella, A. Tscherter, P. Bonsi, N. Sharma, G. Bernardi, D.G. Standaert
Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia
Neurobiol. Dis., 24 (2006), pp. 318-325
Rilstone et al., 2013
J.J. Rilstone, R.A. Alkhater, B.A. Minassian
Brain dopamine-serotonin vesicular transport disease and its treatment
N. Engl. J. Med., 368 (2013), pp. 543-550
Rose et al., 2015
S.J. Rose, X.Y. Yu, A.K. Heinzer, P. Harrast, X. Fan, R.S. Raike, V.B. Thompson, J.F. Pare, D. Weinshenker, Y. Smith, H.A. Jinnah, E.J. Hess
A new knock-in mouse model of l-DOPA-responsive dystonia
Brain, 138 (2015), pp. 2987-3002
Scarduzio et al., 2017
M. Scarduzio, C.N. Zimmerman, K.L. Jaunarajs, Q. Wang, D.G. Standaert, L.L. McMahon
Strength of cholinergic tone dictates the polarity of dopamine D2 receptor modulation of striatal cholinergic interneuron excitability in DYT1 dystonia
Exp. Neurol., 295 (2017), pp. 162-175
Schwarz and Bressman, 2009
C.S. Schwarz, S.B. Bressman
Genetics and treatment of dystonia
Neurol. Clin., 27 (2009), pp. 697-718
vi
Sciamanna et al., 2012
G. Sciamanna, A. Tassone, G. Mandolesi, F. Puglisi, G. Ponterio, G. Martella, G. Madeo, G. Bernardi, D.G. Standaert, P. Bonsi, A. Pisani
Cholinergic dysfunction alters synaptic integration between thalamostriatal and corticostriatal inputs in DYT1 dystonia
J. Neurosci., 32 (2012), pp. 11991-12004
Song et al., 2012
C.H. Song, X. Fan, C.J. Exeter, E.J. Hess, H.A. Jinnah
Functional analysis of dopaminergic systems in a DYT1 knock-in mouse model of dystonia
Neurobiol. Dis., 48 (2012), pp. 66-78
Tepper et al., 2008
J.M. Tepper, C.J. Wilson, T. Koos
Feedforward and feedback inhibition in neostriatal GABAergic spiny neurons
Brain Res. Rev., 58 (2008), pp. 272-281
Thenganatt and Jankovic, 2014
M.A. Thenganatt, J. Jankovic
Treatment of dystonia
Neurotherapeutics, 11 (2014), pp. 139-152
Threlfell and Cragg, 2011
S. Threlfell, S.J. Cragg
Dopamine signaling in dorsal versus ventral striatum: the dynamic role of cholinergic interneurons
Front. Syst. Neurosci., 5 (2011), p. 11
Threlfell et al., 2010
S. Threlfell, M.A. Clements, T. Khodai, I.S. Pienaar, R. Exley, J. Wess, S.J. Cragg
Striatal muscarinic receptors promote activity dependence of dopamine transmission via distinct receptor subtypes on cholinergic interneurons in ventral versus dorsal striatum
J. Neurosci., 30 (2010), pp. 3398-3408
Tolosa and Compta, 2006
E. Tolosa, Y. Compta
Dystonia in Parkinson’s disease
J. Neurol., 253 (Suppl. 7) (2006)
Vii7-13
Yamada et al., 2003
M. Yamada, A.S. Basile, I. Fedorova, W. Zhang, A. Duttaroy, Y. Cui, K.G. Lamping, F.M. Faraci, C.X. Deng, J. Wess
Novel insights into M5 muscarinic acetylcholine receptor function by the use of gene targeting technology
Life Sci., 74 (2003), pp. 345-353
Yorgason et al., 2011
J.T. Yorgason, R.A. Espana, S.R. Jones
Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures
J. Neurosci. Methods, 202 (2011), pp. 158-164
Zhang et al., 2002a
W. Zhang, M. Yamada, J. Gomeza, A.S. Basile, J. Wess
Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1-M5 muscarinic receptor knock-out mice
J. Neurosci., 22 (2002), pp. 6347-6352
Zhang et al., 2002b
W. Zhang, A.S. Basile, J. Gomeza, L.A. Volpicelli, A.I. Levey, J. Wess
Characterization of central inhibitory muscarinic autoreceptors by the use of muscarinic acetylcholine receptor knock-out mice
J. Neurosci., 22 (2002), pp. 1709-1717
Zhang et al., 2009a
L. Zhang, W.M. Doyon, J.J. Clark, P.E. Phillips, J.A. Dani
Controls of tonic and phasic dopamine transmission in the dorsal and ventral striatum
Mol. Pharmacol., 76 (2009), pp. 396-404
Zhang et al., 2009b
T. Zhang, L. Zhang, Y. Liang, A.G. Siapas, F.M. Zhou, J.A. Dani
Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine
J. Neurosci., 29 (2009), pp. 4035-4043
To learn more about the research, grants, and funding at each of the school listed, please click on the photo of the desired university for more information.
|
|
|

|

|

|

|
|
|

|

|

|

|

|