|
Dr. Aparna Wagle Shukla What is the Dystonia Coalition? Dystonia Coalition is an international collaboration between clinical experts, researchers, and patient advocacy groups to advance dystonia research for finding better treatments. The coalition was established in 2009 with the support of the Office of Rare Diseases Research in the National Center for Advancing Translational Sciences and The National Institute of Neurological Disorders and Stroke at the NIH. In the last decade, over 50 sites that provide care to dystonia patients and are interested in advancing research have become members of this coalition. These sites are spread across North America, Europe, Asia, and Australia. In addition to the clinical providers and researchers, there are at least 17 patient advocacy groups committed to the coalition's core activities. The patient advocacy groups play an integral role in identifying the critical needs for the dystonia community from a clinical and scientific perspective. Patient advocates recognize the needs of the community through focused workshops. They work with researchers to design appropriate studies and facilitate patient recruitment to accomplish successful and timely completion. They play an active role in organizing meetings to discuss the annual progress, identify challenges for research, and brainstorm ideas for further advancement of research. Some of the patient advocacy groups are also involved in actively finding and funding dystonia researchers, especially the young and new to the dystonia field. 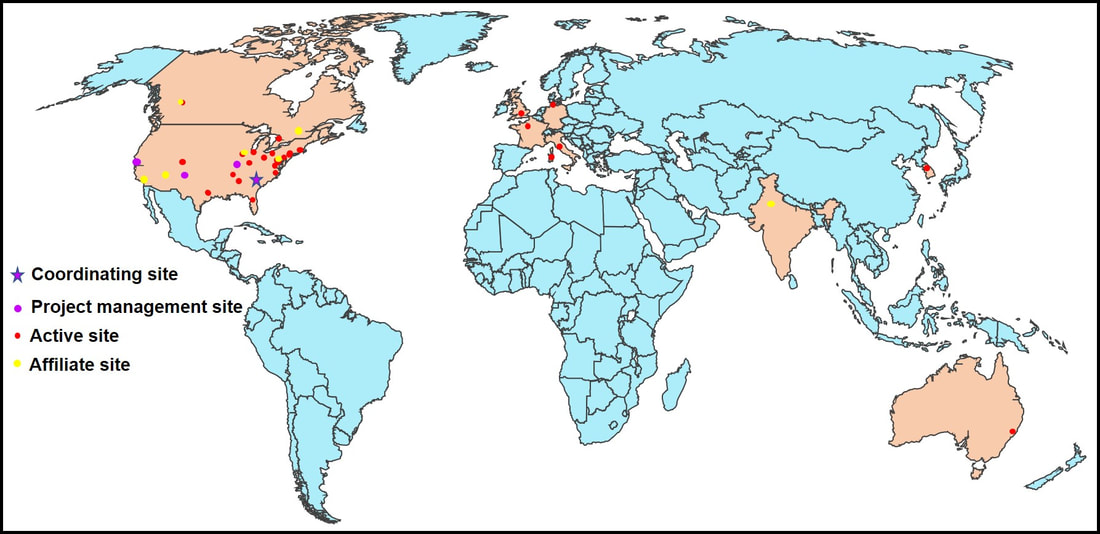 What is the mission of the Dystonia Coalition? Dystonia is a rare disorder which, according to what NIH accepts, is a condition affecting fewer than 200,000 patients in the US. A rare disease poses many unique challenges for the patients, clinicians, and the committed researchers. The patient community may not receive optimal care due to a limited understanding of the disease and the availability of experts. Then the clinicians may not have access to expert guidance or guidelines for management, and the researchers cannot assemble large groups of individuals for participation in clinical studies. Unfortunately, there is also less enthusiasm for funding research for a rare disease. To pursue research in dystonia is further challenging as there is significant heterogeneity. The clinical presentation can vary from one individual to another. The age of onset for symptoms can vary from childhood to adulthood. Some individuals have clear genetic etiology, but the exact cause cannot be ascertained in the vast majority. There is a lack of understanding of natural history for the progression of dystonia symptoms. Currently, we do not have biomarkers that could help in monitoring drug trials for dystonia. Thus, many fundamental challenges can hamper dystonia research. 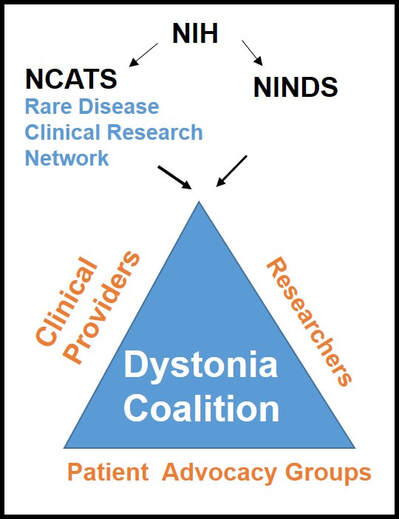 The Dystonia Coalition aims to understand the disease better, develop appropriate tools to monitor patients in clinical trials, and identify biomarkers valuable for clinical research. It has brought hundreds of dystonia providers and thousands of patients to a common platform to facilitate large-scale collaborations to address these critical knowledge gaps. The main ongoing projects for the Coalition include the natural history project, development of diagnostic and rating tools for cervical dystonia, development of similar tools for blepharospasm and laryngeal dystonia, the establishment of objective measures for assessment of symptoms, monitoring outcomes with a patient-centered approach, and building a large biobank through the collection of DNA samples. Their ultimate goal is to accomplish "clinical trial readiness." The current projects are essential for developing effective candidate treatments and ensuring their high likelihood of success. 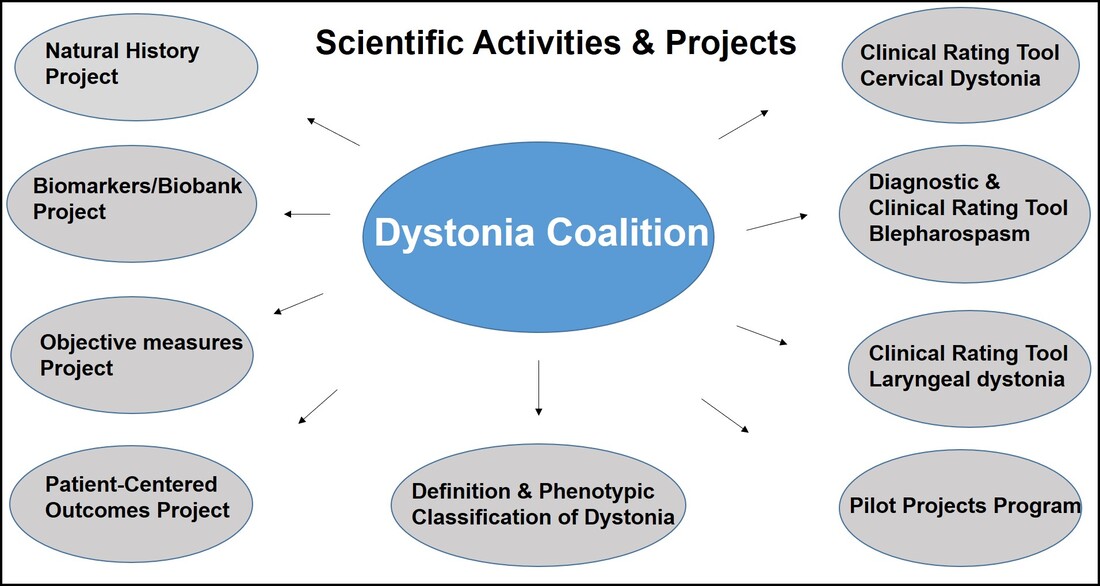 What has the Dystonia Coalition accomplished so far? The efforts of the Dystonia Coalition have led to many significant clinical and scientific achievements. Before the coalition existed, the definition of dystonia had certain missing elements, and the approach for categorizing various forms of dystonia varied among clinicians and researchers. Differences in diagnosis and classification approach invariably led to confusion in the interpretation of many studies. We now have a revised definition for dystonia and a new scheme to classify patients consensually agreed upon by an international team of experts and patient advocates. Then over the last decade, many multicenter projects have been completed successfully. These projects have contributed to understanding the evolution patterns for specific forms of dystonia, the determination of clinically employable measurement scales, and the development of objective tools which could be more sensitive for monitoring clinical symptoms. Due to a broad collaboration network, clinical data, video recordings, and DNA samples for over 3000 patients are available for sharing with researchers worldwide. We can characterize the symptoms better, assess the risk of clinical spread of motor symptoms, recognize the importance of co-occurring nonmotor symptoms commonly, understand the treatment patterns, and identify factors that drive a positive response to the available medical and surgical treatments. Many new young investigators have now joined the dystonia field. These efforts are indeed impressive and commendable. How does the Tyler’s Hope Foundation and the Dystonia Coalition connect? The Tyler’s Hope Foundation and the Dystonia Coalition have common goals. They both engage in keeping the health care providers and the researchers well informed on the needs of the dystonia community. They both are interested and involved in finding financial and non-financial resources for the dystonia researchers. They both aim to promote awareness and education for dystonia through meetings and webinars. The Tyler’s Hope Foundation focuses on genetic dystonia, mainly DYT-Tor1A dystonia. To bring together patients with this genetic diagnosis and guide clinical studies for these patients, Tyler’s Hope maintains a registry of DYT-Tor1A dystonia patients, Dystonia International Patient Registry. On the other hand, the Dystonia Coalition that focuses on all forms of dystonia, is involved with the Global Dystonia Registry to support future clinical trials. As a patient advocacy group, the Tyler’s Hope Foundation is closely supporting the Dystonia Coalition efforts. Together these organizations plan to advance the pace of clinical and translational research to find better treatments and ultimately a cure for dystonia. 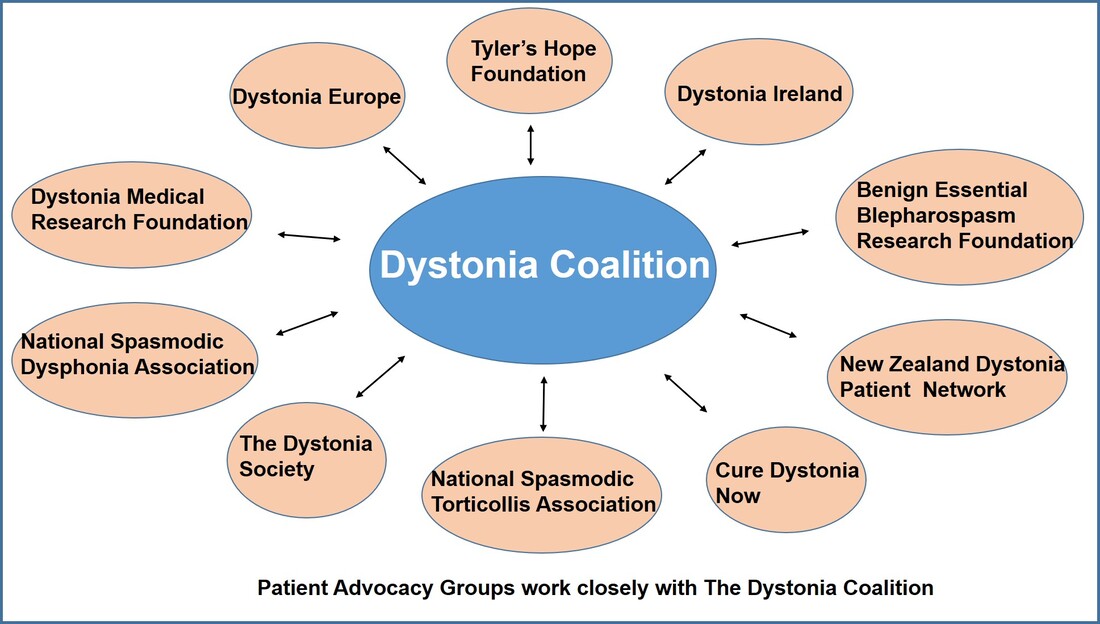 The Dystonia Coalition: A Multicenter Network for Clinical and Translational Studies
Gamze Kilic-Berkmen, Laura J. Wright, Joel S. Perlmutter, Cynthia Comella, Mark Hallett, Jan Teller, Sarah Pirio Richardson, David A. Peterson, Carlos Cruchaga, Codrin Lungu and H. A. Jinnah. Front. Neurol., 08 April 2021 https://doi.org/10.3389/fneur.2021.660909 Dr. Aparna Wagle-Shukla Dystonia is the third most common type of movement disorder. Dystonia occurs due to abnormal firing of brain signals that cause excessive and involuntary or out-of-control muscle contractions. As a result, patients develop abnormal twisting postures, involuntary movements, and lose motor control. Dystonia can affect muscles in either a single or multiple body regions. Examples of such regions include eyelids, face, jaws, neck, vocal cords, torso, limbs, hands, feet, or the entire body. For example, in some individuals with a genetic mutation in a gene called ‘DYT-TOR1A,’ the whole body is commonly affected, and such dystonia is referred to as generalized dystonia. How is dystonia managed? Focal symptoms in a single body region are usually controlled with oral medications and/or botulinum toxin injection treatments. However, when dystonia is generalized, a significant majority of patients require additional surgical treatments. Many times, dystonia is accompanied by other motor symptoms such as tremor, parkinsonism, and myoclonus. Recently, dystonia has been found to affect patients’ mood, sleep, cognition, and balance, referred to as nonmotor symptoms. Therefore, many patients require multidisciplinary care to treat the numerous aspects of dystonia symptomatology successfully. What oral medications are available for treating dystonia? Oral medications for dystonia control the abnormal firing of brain signals. These medications modulate the functions of brain chemicals such as acetylcholine, gamma-aminobutyric acid, or GABA, glutamate, and dopamine that are usually involved in the signaling process. Animal models have shown acetylcholine signaling involving muscarinic receptors in the basal ganglia neurons is abnormal in dystonia. These neurons are located at the base of the brain. In a knock-in mouse model, drugs blocking these receptors (anticholinergic) were observed to reduce dystonia. Medications blocking acetylcholine effects at the muscarinic receptors have been among the oldest treatments available in clinical practice. Trihexyphenidyl (Artane) and benztropine (Cogentin) are commonly prescribed anticholinergic medications and have been observed to effectively treat dystonia symptoms. However, these two drugs non-selectively block all types of muscarinic receptors in the brain (M1, M2, M3, M4, and M5). Thus, side effects related to off-target stimulation such as confusional behavior and constipation are major limiting factors. Scientists have found that selectively blocking M4 receptors, which are highly concentrated in the basal ganglia neurons, will likely result in better clinical outcomes. These selective drugs, with potentially greater benefits and fewer side effects, are currently in the development phase. They will need to be tested and proven to be beneficial in clinical trials before they can be prescribed in clinical practice 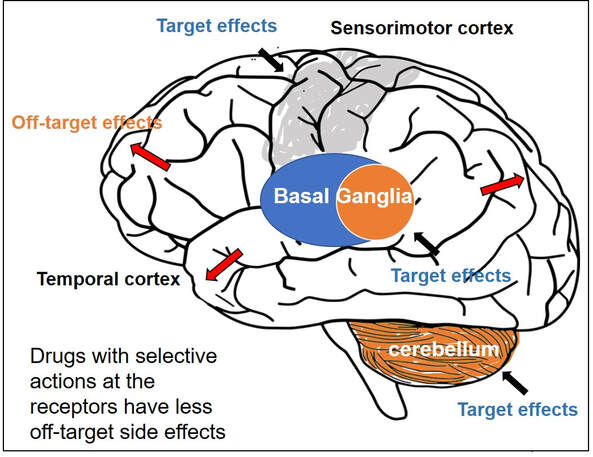 Drugs targeting GABAergic receptors, including benzodiazepines (clonazepam, diazepam, and lorazepam) and baclofen, are other commonly used agents for treating dystonia. GABA is an inhibitory neurotransmitter. An increase in GABA signaling leads to the alleviation of excessive muscle contractions. A recent survey involving more than 2000 dystonia patients found benzodiazepines were the most common oral drugs prescribed for individuals with generalized dystonia, whereas botulinum toxin injections (another powerful muscle relaxant) were most frequently employed for treating focal dystonia. Zolpidem is a medication that also modulates GABA receptors in a slightly different way. It was used in a small study of 34 patients with focal dystonia. The study found significant improvements, but further research is needed on whether the clinical benefits can be extended to generalized dystonia, such as the DYT-TOR1A dystonia. Dopamine and dystonia have a complex relationship as medications that increase dopamine or decrease dopamine can improve clinical symptoms in certain forms of dystonia. Drugs such as levodopa-carbidopa, pramipexole, and ropinirole increase dopamine signaling, and drugs like tetrabenazine and deutetrabenazine decrease dopamine signaling through dopamine depletion. Drug development can be an extremely lengthy and costly process. Thus, many clinical trials repurpose drugs to leverage their pharmacological properties for benefiting a broader range of clinical applications. One example is escitalopram (Lexapro), a selective serotonin reuptake inhibitor, a drug frequently used to treat pain and depression and generally well tolerated was recently found to improve cervical dystonia symptoms. The benefits were observed to be independent of their influence on mood circuitry. Another example is Perampanel, a drug antagonizing the action of glutamate neurotransmitter and approved for the treatment of epilepsy. Perampanel was recently tested in focal dystonia as the target receptors are widely distributed in the basal ganglia neurons. The target receptors are also seen in other neurons like the cerebellar neurons important for dystonia. While the drug was safe and tolerated by most patients, further studies are needed to confirm clinical efficacy. 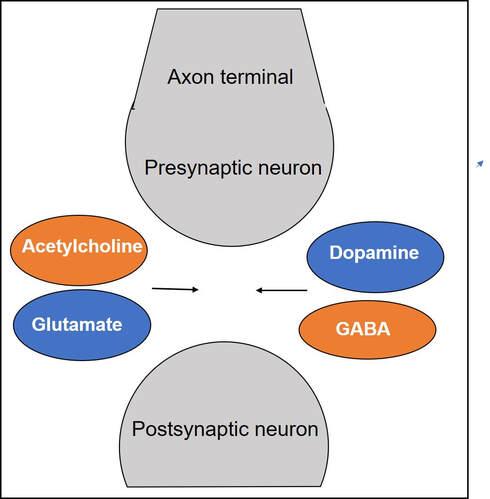 What are the general principles for using oral medications?
Oral medications should be initiated at low doses and titrated up slowly to avoid off-target side effects, especially in older patients. Sometimes for certain medications like anticholinergic drugs, peak benefits are observed only weeks after maintaining constant doses. If the peak benefits are not adequate or are short-lasting, a further buildup of doses is recommended. If dose-limiting side effects develop with further escalation, an additional line of therapy is needed to achieve the best possible symptom control. Are treatments available if a specific cause or etiology is identified? In some individuals with dystonia, a specific cause can be identified. For example, when dystonia occurs due to a deficiency of enzymes needed to synthesize dopamine, the replacement of dopamine is found to alleviate symptoms (dopa-responsive dystonia). Sometimes dystonia is related to abnormalities in protein or lipid metabolism or deposition of heavy metals in the brain (e.g., copper, iron). In these circumstances, once the diagnosis is confirmed, appropriate specific therapies can be initiated. References Update on current and emerging therapies for dystonia; Karlo J Lizarraga, Duha Al-Shorafat & Susan Fox. Neurodegener. Dis.Manag. (2019) 9(3), 135–147 Anticholinergic drugs rescue synaptic plasticity in DYT1 dystonia: role of M1 muscarinic receptors. Maltese M, Martella G, Madeo G et al Mov. Disord. 29(13), 1655–1665 (2014 Dystonia treatment: patterns of medication use in an international cohort. Pirio Richardson S, Wegele AR, Skipper B, Deligtisch A, Jinnah HA. Neurology 88(6), 543–550 (2017). Discovery of the first selective M4 muscarinic acetylcholine receptor antagonists with in vivo anti-parkinsonian and anti-dystonic efficacy. Mark S. Moehle, Aaron M. Bender, Jonathan W. Dickerson, et al. doi: https://doi.org/10.1101/2020.10.12.324152  Who is Cristopher Bragg?  DR. CRISTOPHER BRAGG Dr. Cristopher Bragg is an Assistant Professor of Neurology at Massachusetts General Hospital and Harvard Medical School. His lab studies cellular mechanisms underlying hereditary dystonias, with a current focus on three forms in particular: DYT1 dystonia, DYT6 dystonia, and X-Linked Dystonia-Parkinsonism (XDP). He also serves as the Director of the MGH Collaborative Center for X-Linked Dystonia-Parkinsonism (CCXDP), an international consortium focused exclusively on a rare syndrome that is endemic to the Philippines. What are DYT1, DYT6, and XDP?  THAP1 DYT-6 MOLECULAR STRUCTURE DYT1 and DYT6 are both generalized dystonias that typically develop in childhood or adolescence. They are often designated as “isolated” or “pure” dystonias, because in most patients, dystonia is the sole or primary symptom. XDP, in contrast, usually arises in adulthood, most often during the third or fourth decade of life. Unlike DYT1 and DYT6, XDP combines features of both dystonia and parkinsonism often in a temporal progression. The pattern that has been documented most frequently consists of an initial focal dystonia that spreads to other body regions— as parkinsonian symptoms also emerge over time. Some patients, however, may exhibit parkinsonian signs as the first symptom and experience a milder course of disease progression,— and the reasons for this heterogeneity are not yet understood. What causes these disorders?  XANDRA BREAKEFIELD PHD The genetic cause of DYT1 was discovered in 1997 by Drs. Xandra Breakefield and Laurie Ozelius at Massachusetts General Hospital. It was the culmination of many years of painstaking work that took place long before the human genome was sequenced, when the methods for finding disease-related genes were still in their infancy. They found that DYT1 patients had a specific deletion in a small stretch of the TOR1A gene, which encodes a protein termed torsinA. Since that first report, a small number of additional mutations in TOR1A have been found, but the vast majority of DYT1 patients all appear to share the same original gene variant.  LAURIE OZELIUS PHD DYT6 dystonia is associated with mutations in the THAP1 gene. Unlike DYT1, which is caused by the same gene deletion in nearly all cases, DYT6 has been linked to a large number of different mutations in THAP1. It is not yet known if these different mutations account for differences in symptoms, as so far there are no clear relationships between variants in THAP1 and patterns of clinical disease. While cases of DYT1 and DYT6 have been found in multiple patient populations around the world, XDP has arisen uniquely among individuals with ancestry to the island of Panay in the Philippines. It is caused by the insertion of a long stretch of DNA called a “retrotransposon.” Retrotransposons come in several varieties, many of which have been derived from ancient viruses that deposited their genetic content into our genomes millions of years ago. They are considered “mobile DNA” because of their ability to move from one chromosome to another by reinserting copies of themselves into other genomic locations - a discovery first made by Dr. Barbara McClintock for which she was awarded the Nobel Prize in Medicine in 1983. All living organisms contain various forms of mobile DNA. In humans, these elements are scattered throughout our genomes; most are completely benign and have lost their ability to move over the course of evolution. However, a few types of retrotransposons are still active, and within the Panay population, one such example has become inserted into a gene called TAF1, thereby disrupting its function and causing XDP.  BREAKEFIELD LABORATORY Why study these syndromes together?  XDP DYSTONIA WHICH IS VERY COMMON ON THE ISLAND OF PANAY Despite the differences between DYT1, DYT6, and XDP, the proteins that are affected in these syndromes play inter-related roles within cells. The DYT6 protein, THAP1, and XDP protein, TAF1, both reside within the cell’s nucleus and regulate how genes are turned on or off, a process known as transcription. The DYT1 protein, torsinA, acts within the adjacent envelope that surrounds the nucleus and is important for its shape and organization. Numerous research studies over many years have characterized how torsinA functions within the nuclear envelope and how the mutation that causes DYT1 dystonia may compromise this cellular compartment. Less is known about THAP1 and TAF1, but recent studies are now revealing how transcription in DYT6 and XDP cells is affected by the dystonia-related variants in these genes.
The Bragg lab studies these and other dystonic syndromes together by analyzing patient cells and characterizing their patterns of gene transcription, which can reveal cellular pathways that are affected in these cells. The overall goal of this work is to look for any defects that may be common to multiple forms of dystonia. If such deficits exist, then they may represent potential targets for designing new therapies that would be of broad benefit to patients with dystonia. Dr. Edgar Rodriguez weighs in on Gene Therapy for Brain Diseases: A Path for DYT-1 Dystonia6/3/2020
 SCIWORTHY.COM (PICTURE) WHICH HAS GREAT IMAGES AND A GREAT SUMMARY WEBSITE OF THIS TOPIC! Gene Therapy for DYT-1 Dystonia and other diseases? We sat down with one the experts on gene therapy for DYT-1 dystonia and other central nervous system diseases and we picked his brain on this topic. The interview was fascinating! Who is Edgar Rodriguez?  Dr. Edgardo (Edgar) Rodríguez-Lebrón has 20 years of experience in the gene therapy space with a focus on diseases of the nervous system. He has engineered and developed gene-based therapies to prevent or reverse neurodegenerative processes caused by the accumulation of toxic mutant proteins. He is currently Scientific Advisor to Tyler's Hope, Chief Science Officer of Andante Biologics and a co-founder of early-stage biotech companies developing molecular therapies for CNS diseases. He holds an Adjunct Faculty appointment at the University of Florida where he trained as a graduate student. Go Gators! Often, diseases that affect the brain are caused by genetic mutations that prevent the normal function of a gene. In other cases, genetic mutations can cause a gene to ‘gain’ a new function that is toxic to the brain. Gene therapies are a class of medicines designed to specifically (i) replace a missing genetic function, (ii) block a ‘gained’ toxic genetic function, or (iii) limit or enhance cellular pathways to support overall brain function.  What is a viral-based gene therapy? 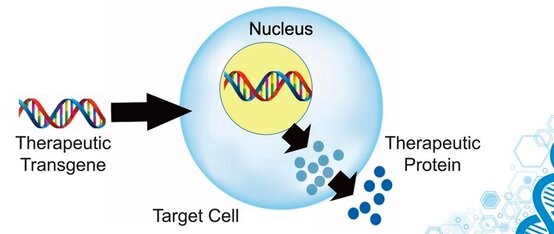
What makes AAV a desirable vector for CNS-targeted gene therapies? 
What are some recent major developments in AAV CNS gene therapy?  Recent developments include:
What are some of the major hurdles still being faced by AAV-based gene therapies?  Hurdles include:
How can an AAV-based gene therapy be used in DYT1 dystonia?  Applications to DYT-1:
What is on the horizon for AAV-based gene therapies as it applies to DYT1?  On the horizon:
 COVID-19 and Dystonia Care: What do you need to know? While we patiently wait for the COVID-19 crisis to resolve, we approached leading dystonia experts for tips and advice on caring for patients. We were fortunate to sit down with one of the leading clinical and research experts, neurologist Dr. Aparna Wagle Shukla. She shared with us her “top 5” tips for dystonia care during the COVID crisis. Who is Dr. Aparna Wagle Shukla? 
 What should every dystonia patient know about the Corona virus?
 THERE IS NO VACCINE FOR COVID AT THE CURRENT TIME WHICH MEANS PREVENTION IS THE KEY.
 What are important precautions every dystonia patient should take at home?

 How important is social distancing for dystonia patients? What is the risk of COVID-19 in dystonia?


 What should a dystonia patients know about medication management during the COVID-19 crisis? What about the need to see a doctor during the COVID crisis?
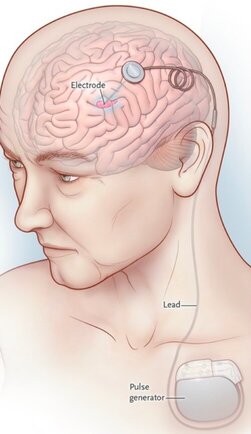 What do you tell dystonia patients with deep brain stimulation (DBS) devices during the COVID-19 outbreak?
 There are so many exciting projects going on in dystonia research that it has become hard to keep up with all of the advances. We sat down with one of the worlds top dystonia and Parkinson imaging researchers (Dr. David Vaillancourt) to talk about the “top 5” updates in therapies and in dystonia imaging research. Who is David Vaillancourt? 
 1) What is imaging and how is it used in dystonia?
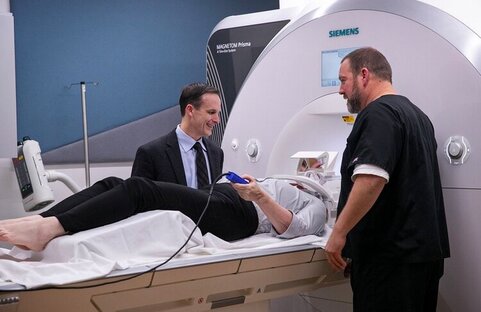 2) What is a specific example in humans for how imaging is being used in dystonia?
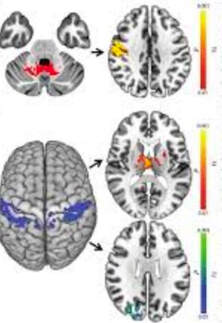 3) What is a specific example in animals for how imaging is being used in dystonia?
 4) How can imaging be used in humans to assess a new therapeutic (e.g. drug, device)?
 5) What does this all mean for people with dystonia?
|
Archives
September 2021
Categories |


 RSS Feed
RSS Feed
Location:
|